GEO生信数据挖掘(八)富集分析(GO 、KEGG、 GSEA 打包带走)
第六节,我们使用结核病基因数据,做了一个数据预处理的实操案例。例子中结核类型,包括结核,潜隐进展,对照和潜隐,四个类别。第七节延续上个数据,进行了差异分析。 本节对差异基因进行富集分析。
目录
数据展示
GO富集分析 -对基因名称映射基因ID
GO富集分析 -从org.Hs.eg.db库中去匹配基因
KEGG富集分析 (不详细讲了看注释)
GSEA 富集分析
更多复杂的图(关联网络图、八卦图 、弦图)
数据展示
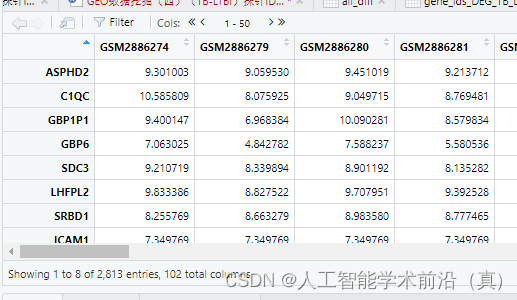
差异基因计算完毕的指标如下图所示

差异基因筛选后表达矩阵
GO富集分析 -对基因名称映射基因ID
加载数据
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&加载数据&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
load( "DEG_TB_LTBI_step13.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&加载数据&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&library(clusterProfiler)
library(org.Hs.eg.db)#增加基因名
all_diff$SYMBOL=rownames(all_diff)#基因名称转换注释
gene_ids_DEG_TB_LTBI = bitr(geneID = rownames(dataset_TB_LTBI_DEG),fromType="SYMBOL",toType = c("ENTREZID","ENSEMBL","SYMBOL"),OrgDb = 'org.Hs.eg.db',drop =TRUE)#合并 增加logFC 为后续GSEA富集分析所需数据准备
gene_ids_DEG_TB_LTBI <- merge(gene_ids_DEG_TB_LTBI,all_diff,by="SYMBOL")#观察
dim(gene_ids_DEG_TB_LTBI)
head(gene_ids_DEG_TB_LTBI)#获取基因ID ENSEMBL
gene_ENSEMBL <- gene_ids_DEG_TB_LTBI$ENSEMBL
gene_ENTREZID <- gene_ids_DEG_TB_LTBI$ENTREZID
gene_SYMBOL<- gene_ids_DEG_TB_LTBI$SYMBOL
经过映射,2813个差异基因得到2551个基因ID,下图为三种不同形式的基因名称,富集分析时,按需进行转换。

GO富集分析 -从org.Hs.eg.db库中去匹配基因
#Go富集分析,从库中去匹配
go <- enrichGO(gene_SYMBOL,OrgDb = org.Hs.eg.db, ont='ALL',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'SYMBOL')#进行GO富集,确定P值与Q值得卡值并使用BH方法对值进行调整。
#查看富集结果
dim(go)
#导出GO富集的结果
write.csv(go,file="go1.csv")绘制气泡图
#绘制气泡图
pdf(file="15aGO富集分析step15.pdf", width = 9, height = 6)
dotplot(go,showCategory=20,label_format = 80)#气泡图dev.off()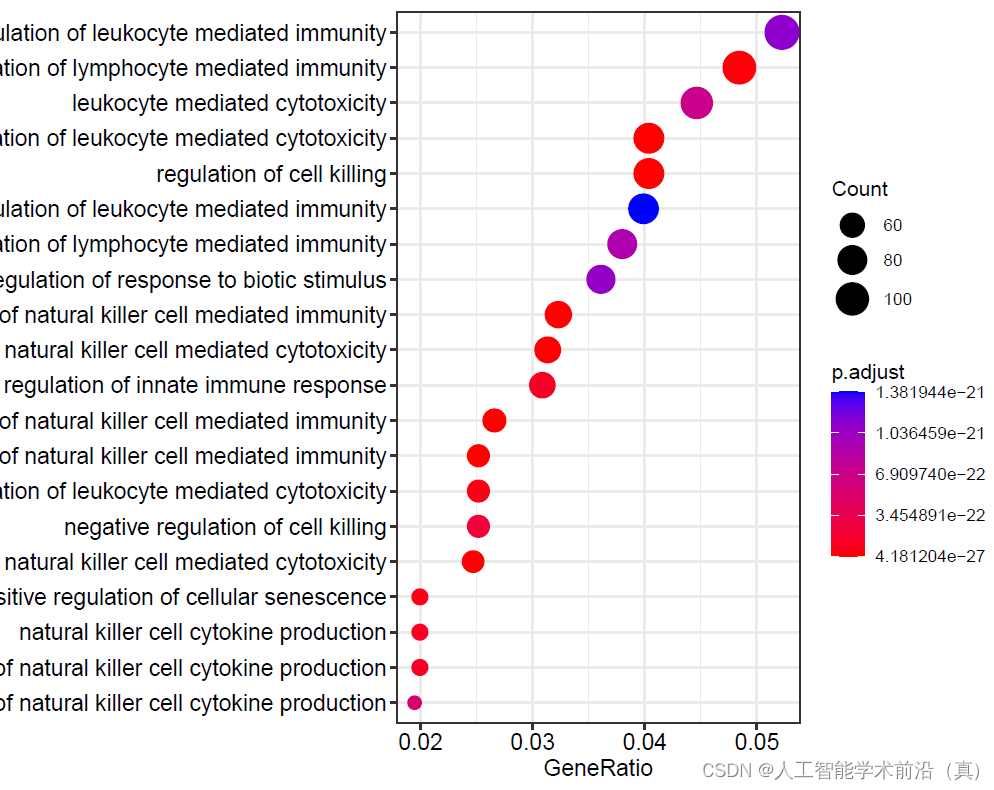
三种不同类别的合并的气泡图(#CC细胞组件,MF分子功能,BP生物学过程)
pdf(file="15bGO富集分析三组step15.pdf", width = 9, height = 6)#CC细胞组件,MF分子功能,BP生物学过程
goCC <- enrichGO(gene = gene_ENTREZID, #基因列表(转换的ID)keyType = "ENTREZID", #指定的基因ID类型,默认为ENTREZIDOrgDb=org.Hs.eg.db, #物种对应的org包ont = "CC", #CC细胞组件,MF分子功能,BP生物学过程pvalueCutoff = 0.05, #p值阈值pAdjustMethod = "fdr", #多重假设检验校正方式minGSSize = 1, #注释的最小基因集,默认为10maxGSSize = 500, #注释的最大基因集,默认为500qvalueCutoff = 0.2, #q值阈值readable = TRUE) #基因ID转换为基因名goBP <- enrichGO(gene_ENTREZID,OrgDb = org.Hs.eg.db, ont='BP',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'ENTREZID')goMF <- enrichGO(gene_ENTREZID,OrgDb = org.Hs.eg.db, ont='MF',pAdjustMethod = 'BH',pvalueCutoff = 0.05, qvalueCutoff = 0.2,keyType = 'ENTREZID')
#通过ggplot2将BP、MF、CC途径的富集结果挑选前8条绘制在一张图上
barplot(go,label_format=100, split="ONTOLOGY")+ facet_grid(ONTOLOGY~.,scale="free")dev.off()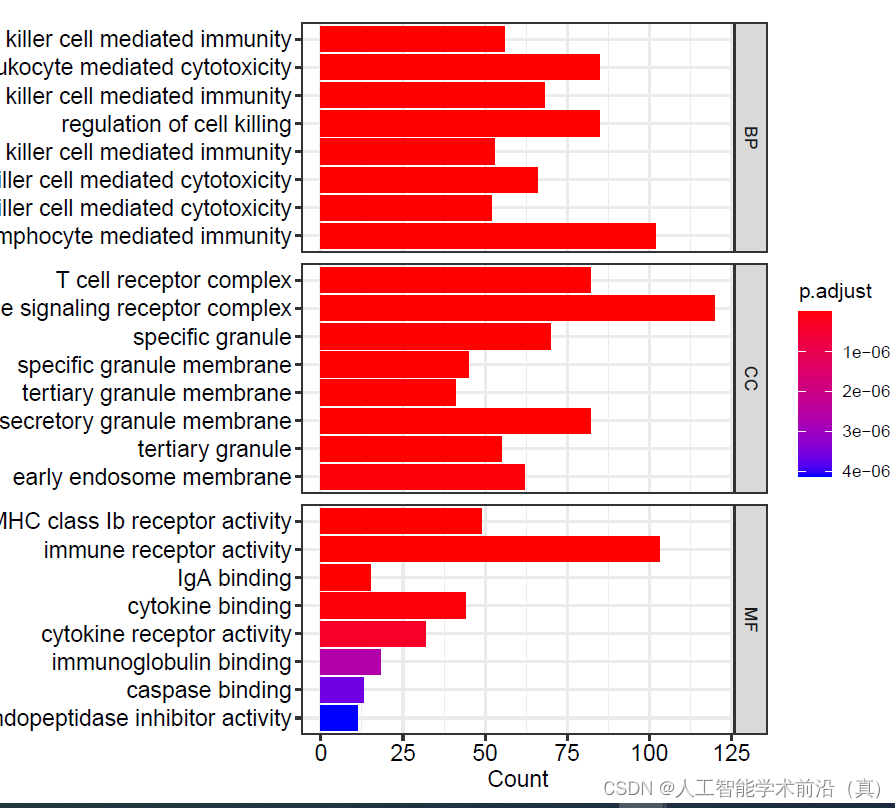
KEGG富集分析 (不详细讲了看注释)
#+=================================================================
#============================================================
#+========KEGG富集分析 气泡图step16===================
#+==========================================
#+================================#KEGG富集分析
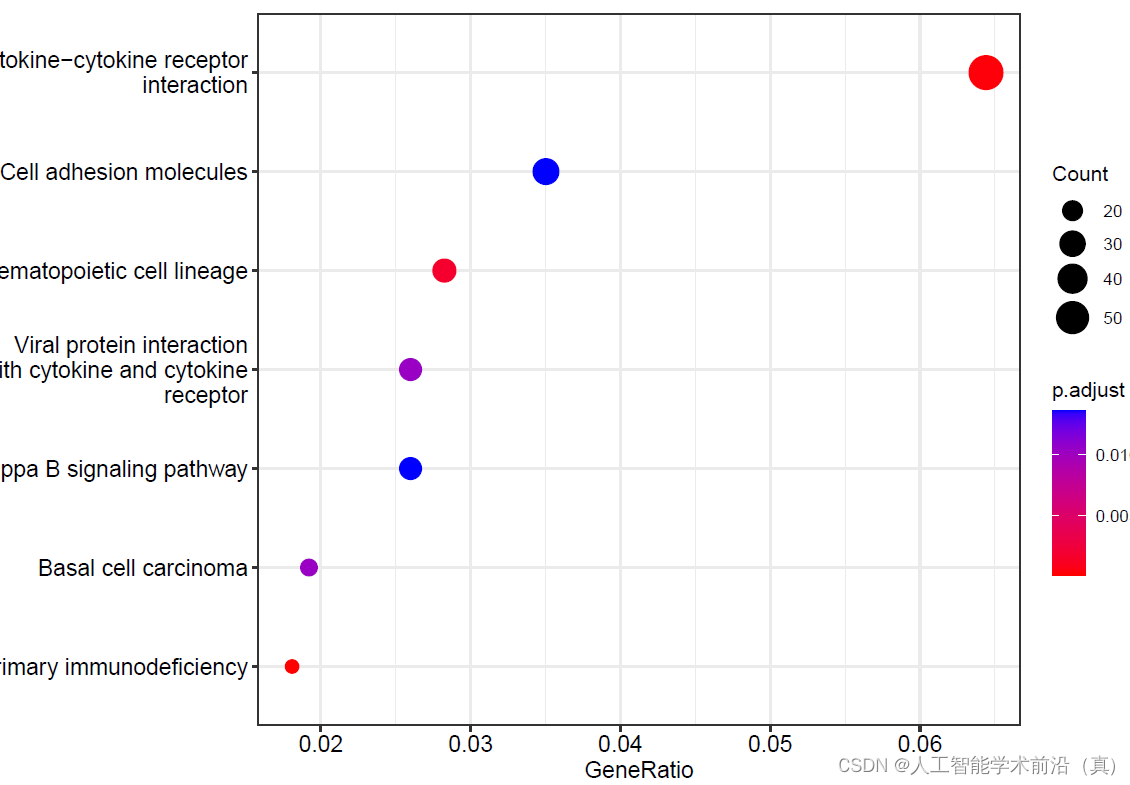
pdf(file="16KEGG富集分析step16.pdf", width = 9, height = 6)
kegg<- enrichKEGG(gene = gene_ENTREZID, #基因列表(ENTREZID ID: 54490,51144,31,3906) organism = "hsa", #物种keyType = "kegg", #指定的基因ID类型,默认为keggminGSSize = 3, maxGSSize = 500,pvalueCutoff = 0.05, pAdjustMethod = "fdr", # pAdjustMethod = 'BH'qvalueCutoff = 0.02)
#观察
dim(kegg)
#绘制气泡图
dotplot(kegg)
dev.off()#kegg 增加可读性,对基因ID 转基因名
kegg_enrich_results <- DOSE::setReadable(kegg, OrgDb="org.Hs.eg.db", keyType='ENTREZID') #ENTREZID to gene Symbol#保存kegg结果
write.csv(kegg_enrich_results@result,'KEGG_gene_up_enrichresults.csv')
#save(kegg_enrich_results, file ='KEGG_gene_up_enrichresults.Rdata')##查看与选择所需通路
kegg_enrich_results@result$Description[1:10] #查看前10通路###选择所需通路的ID号
i=1
select_pathway <- kegg_enrich_results@result$ID[i] #选择所需通路的ID号#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(gene_ids_DEG_TB_LTBI,go,keggfile ="15_gene_ids_DEG_TB_LTBI.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
GSEA 富集分析
#+=================================================================
#============================================================
#+========GSEA 富集分析 气泡图step17===================
#+==========================================
#+================================# GSEA 分析
#需要把多个方法取并集
#该方法的输入需要基因和 logFC 排序后的结果
#不同方法 相同基因的的logFC值不一样,直接保留第一个重复基因library(stringr)## 去重 #去除NA值
dim(gene_ids_DEG_TB_LTBI)
colnames(gene_ids_DEG_TB_LTBI)gene_list_df = gene_ids_DEG_TB_LTBI[,c('ENTREZID','logFC')]
gene_list_df_na <- na.omit(gene_list_df)gene_ids_TB_LTBI_distinct <- dplyr::distinct(gene_list_df_na,ENTREZID,.keep_all=TRUE) dim(gene_ids_TB_LTBI_distinct)
gene_list=gene_ids_TB_LTBI_distinct$logFC #提取logFC列
names(gene_list)=gene_ids_TB_LTBI_distinct$ENTREZID #加上ENTREZID
gene_list_gsea = sort(gene_list, decreasing = T) #降序排列gsea_KEGG <- gseKEGG(gene_list_gsea,organism = "hsa",keyType = "kegg")gsea_KEGG_d <- as.data.frame(gsea_KEGG)gsea_KEGG_d
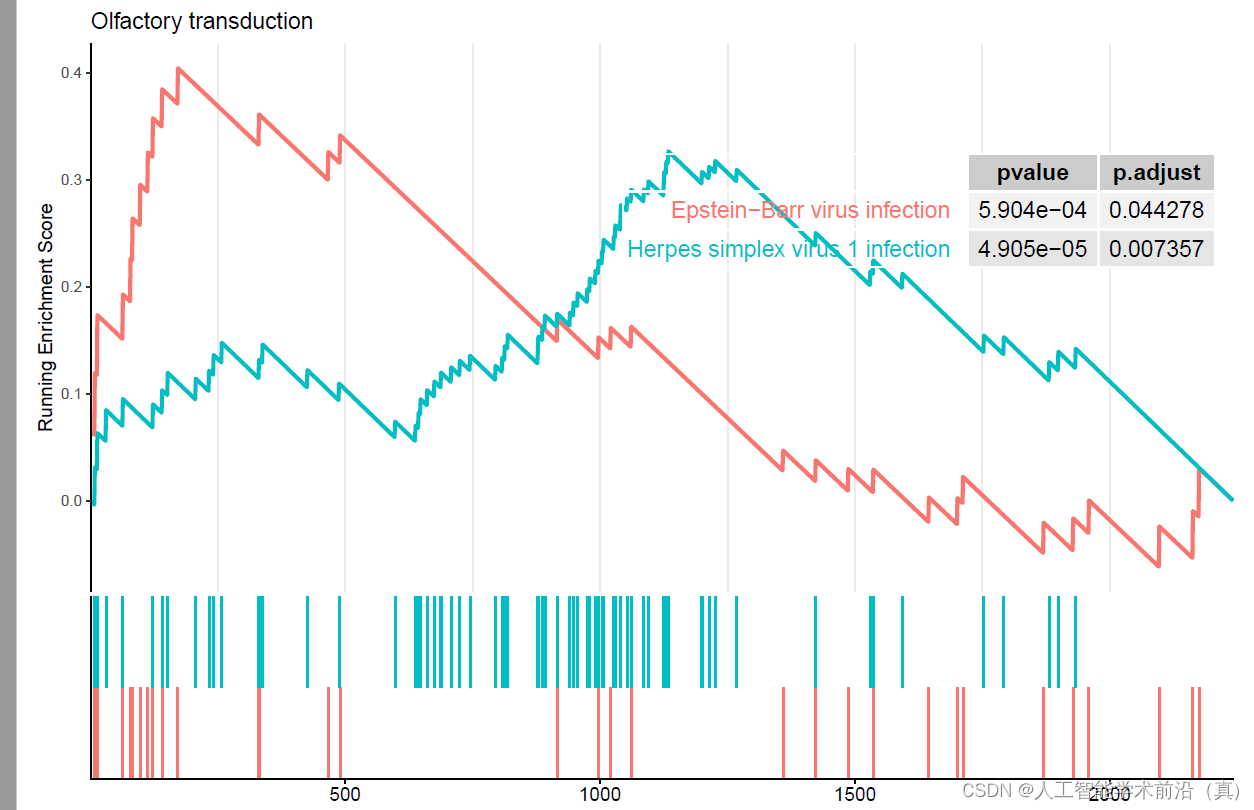
#path 为需要展示的pathway id,这里展示的是enrichment score最高的4条通路t_index=order(gsea_KEGG_d$enrichmentScore,decreasing = T)
path=rownames(gsea_KEGG[t_index,]) #选择展示的 pathwayrownames(gsea_KEGG[t_index,]) [1:4]#作图
pdf(file="17GSEA富集分析step17.pdf", width = 9, height = 6)
gseaplot2(gsea_KEGG,path, subplots = 1:2, #展示前2个图pvalue_table = T, #显示p值title = "Olfactory transduction", #设置titlebase_size = 10, #字体大小color="red") #线条颜色可选
dev.off()#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
save(gene_ids_DEG_TB_LTBI,go,kegg,gene_list_gsea,gsea_KEGG,file ="17_gene_ids_DEG_TB_LTBI.Rdata")
#++++++++++++++++++++++++++++++++++++++++++++++++++++++++++++
#+&&&&&&&&&&&&&&&&&&数据保存&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
#&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&&
更多复杂的图(关联网络图、八卦图 、弦图)
参考 最全的GO, KEGG, GSEA分析教程(R),你要的高端可视化都在这啦![包含富集圈图] - 糖糖家的老张的文章 - 知乎 https://zhuanlan.zhihu.com/p/377356510
原文链接:https://blog.csdn.net/qq_50898257/article/details/120588222
#+=================================================================
#============================================================
#+========富集分析 更多的图step18===================
#+==========================================
#+================================
library(clusterProfiler)
library(enrichplot)
#+富集基因与所在功能集/通路集的关联网络图:
enrichplot::cnetplot(go,circular=FALSE,colorEdge = TRUE)#基因-通路关联网络图
enrichplot::cnetplot(kegg,circular=FALSE,colorEdge = TRUE)#circluar为指定是否环化,基因过多时建议设置为FALSEGO2 <- pairwise_termsim(go)
KEGG2 <- pairwise_termsim(kegg)
enrichplot::emapplot(GO2,showCategory = 50, color = "p.adjust", layout = "kk")#通路间关联网络图
enrichplot::emapplot(KEGG2,showCategory =50, color = "p.adjust", layout = "kk")write.table(kegg$ID, file = "KEGG_IDs.txt", #将所有KEGG富集到的通路写入本地文件查看append = FALSE, quote = TRUE, sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")
#打印几条通路名称看看
kegg$ID[1:3]
#打开浏览器观察通路
browseKEGG(kegg,"hsa04660")#选择其中的hsa05166通路进行展示#富集弦图
genedata<-data.frame(ID=gene_ids_DEG_TB_LTBI$SYMBOL ,logFC=gene_ids_DEG_TB_LTBI$logFC)
write.table(go$ONTOLOGY, file = "GO_ONTOLOGYs.txt", #将所有GO富集到的基因集所对应的类型写入本地文件从而得到BP/CC/MF各自的起始位置如我的数据里是1,2103,2410append = FALSE, quote = TRUE, sep = " ",eol = "\n", na = "NA", dec = ".", row.names = TRUE,col.names = TRUE, qmethod = c("escape", "double"),fileEncoding = "")'''
根据计算出的go 文件数量,调整
'''
GOplotIn_BP<-go[1:178,c(2,3,7,9)] #提取GO富集BP的前10行,提取ID,Description,p.adjust,GeneID四列
GOplotIn_CC<-go[179:194,c(2,3,7,9)]#提取GO富集CC的前10行,提取ID,Description,p.adjust,GeneID四列
GOplotIn_MF<-go[195:209,c(2,3,7,9)]#提取GO富集MF的前10行,提取ID,Description,p.adjust,GeneID四列library(stringr)
GOplotIn_BP$geneID <-str_replace_all(GOplotIn_BP$geneID,'/',',') #把GeneID列中的’/’替换成‘,’
GOplotIn_CC$geneID <-str_replace_all(GOplotIn_CC$geneID,'/',',')
GOplotIn_MF$geneID <-str_replace_all(GOplotIn_MF$geneID,'/',',')
names(GOplotIn_BP)<-c('ID','Term','adj_pval','Genes')#修改列名,后面弦图绘制的时候需要这样的格式
names(GOplotIn_CC)<-c('ID','Term','adj_pval','Genes')
names(GOplotIn_MF)<-c('ID','Term','adj_pval','Genes')
GOplotIn_BP$Category = "BP"#分类信息
GOplotIn_CC$Category = "CC"
GOplotIn_MF$Category = "MF"BiocManager::install('GOplot')
library(GOplot)
circ_BP<-GOplot::circle_dat(GOplotIn_BP,genedata) #GOplot导入数据格式整理
circ_CC<-GOplot::circle_dat(GOplotIn_CC,genedata)
circ_MF<-GOplot::circle_dat(GOplotIn_MF,genedata) chord_BP<-chord_dat(data = circ_BP,genes = genedata) #生成含有选定基因的数据框
chord_CC<-chord_dat(data = circ_CC,genes = genedata)
chord_MF<-chord_dat(data = circ_MF,genes = genedata)
'''
> chord_CC<-chord_dat(data = circ_CC,genes = genedata)
Error in `[<-`(`*tmp*`, g, p, value = ifelse(M[g] %in% sub2$genes, 1, : subscript out of bounds我去检查了go和genelist的数据结构发现,genelist里的gene用的是gene名,而go里的基因用的是基因ID,不一样了,所以跑不出结果,所以我把genelist的gene换成了基因ID,就能跑出来了。作者:ff的小世界勿扰
链接:https://www.jianshu.com/p/ee4012fd253f
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
'''#可以画 数量太多了
GOChord(data = chord_BP,#弦图title = 'GO-Biological Process',space = 0.01,#GO Term间距limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,lfc.col = c('red','white','blue'), #上下调基因颜色process.label = 10) #GO Term字体大小
GOChord(data = chord_CC,title = 'GO-Cellular Component',space = 0.01,limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,lfc.col = c('red','white','blue'), process.label = 10)
GOChord(data = chord_MF,title = 'GO-Mollecular Function',space = 0.01,limit = c(1,1),gene.order = 'logFC',gene.space = 0.25,gene.size = 5,lfc.col = c('red','white','blue'), process.label = 10)
'''
Warning messages:
1: Using size for a discrete variable is not advised.
2: Removed 15 rows containing missing values (`geom_point()`).
'''富集分析完毕!
回顾我们用到方法,差异分析后进行富集分析,理论基础实际上就是简单的找不同,分析。
实际应用种,由于基因之间存在关联,另一套分析理论考虑的是基因之间的相互作用,下节,我们来看非常火的WGCNA 共表达加权网络进行基因分析。
相关文章:
GEO生信数据挖掘(八)富集分析(GO 、KEGG、 GSEA 打包带走)
第六节,我们使用结核病基因数据,做了一个数据预处理的实操案例。例子中结核类型,包括结核,潜隐进展,对照和潜隐,四个类别。第七节延续上个数据,进行了差异分析。 本节对差异基因进行富集分析。 …...

高校教务系统登录页面JS分析——华南理工大学
高校教务系统密码加密逻辑及JS逆向 本文将介绍高校教务系统的密码加密逻辑以及使用JavaScript进行逆向分析的过程。通过本文,你将了解到密码加密的基本概念、常用加密算法以及如何通过逆向分析来破解密码。 本文仅供交流学习,勿用于非法用途。 一、密码加…...

人工智能之PyTorch数据操作-Python版
PyTorch数据操作 # 导入PyTorch import torch [张量表示一个由数值组成的数组,这个数组可能有多个维度]。 具有一个轴的张量对应数学上的向量(); 具有两个轴的张量对应数学上的矩阵(matrix);…...

星环科技向量数据库Transwarp Hippo1.1发布:一库搞定向量+全文联合检索,提升大模型准确率
星环科技向量数据库Transwarp Hippo自发布已来,受到了众多用户的欢迎,帮助用户实现向量数据的存储、管理和检索,探索和实践大模型场景。在与用户不断地深入交流以及实践中,Hippo迎来了V1.1版本,一套系统即可支持向量与全文联合检索,提高文本数据的召回精度,从而提升大语…...
理解LoadRunner,基于此工具进行后端性能测试的详细过程(下)
5、录制并增强虚拟用户脚本 从整体角度看,用LoadRunner 开发虚拟用户脚本主要包括下面四步骤: 识别测试应用使用的协议 录制脚本 完善录制得到的脚本 验证脚本的正确性 识别被测应用使用的协议 如果明确知道了被测系统所采用的协议,可…...
使用和理解说明)
K8s上的监控系统(Grafana)使用和理解说明
Grafana (集成Prometheus On K8s集成)主要步骤说明 客户端指标收集 —— K8s 集群资源等 —— Prometheus 监控数据收集 —— Grafana —— 通过PromQL 进行数据查询 —— 预警告警等通知 Kubernetes集群资源:这包括了CPU、内存、磁盘、网络等各种类型的资源。这些资…...

【netty从入门到放弃】netty转发tcp数据到多客户端
目录 创建数据库表xml实体类启动类线程类客户端代码handlecontroller类缓存tcp链接 接到一个需求,需要实现转发通讯模块tcp数据其他的服务器,也就是转发tcp数据到多客户端 任务拆解: 首先需要建立多客户端,每个客户端有一个独立的clientId和…...
Linux | gdb的基本使用
目录 前言 一、调试文件的生成 二、调试指令 1、选择调试文件 2、查看代码 3、运行代码 4、断点 5、打印与常显示 6、其他 总结 前言 前面我们学习了如何使用gcc/g来进行对代码进行编译,本章我们将使用gdb来对代码进行调试,学习本章的前提是有…...
C++之this指针
前言 C中对象模型和this指针是面向对象编程中的重要概念。对象模型描述了对象在内存中的布局和行为,包括成员变量、成员函数的存储方式和访问权限。this指针是一个隐含的指针,指向当前对象的地址,用于在成员函数中引用当前对象的成员变量和成…...
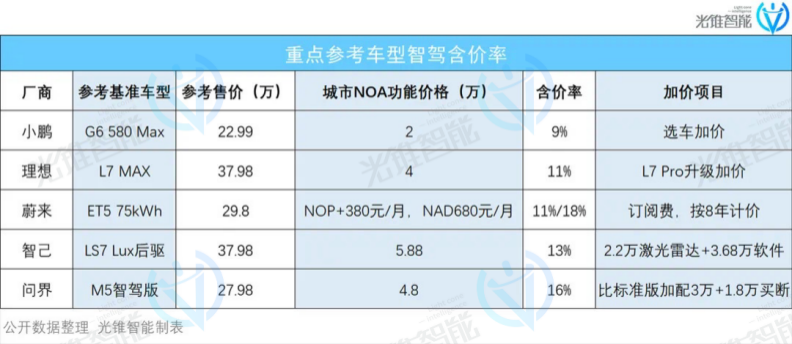
大模型,重构自动驾驶
文|刘俊宏 编|王一粟 大模型如何重构自动驾驶?答案已经逐渐露出水面。 “在大数据、大模型为特征,以数据驱动为开发模式的自动驾驶3.0时代,自动驾驶大模型将在车端、云端上实现一个统一的端到端的平台管理。”毫末智…...
Jmeter执行接口自动化测试-如何初始化清空旧数据
需求分析: 每次执行完自动化测试,我们不会执行删除接口把数据删除,而需要留着手工测试,此时会导致下次执行测试有旧数据我们手工可能也会新增数据,导致下次执行自动化测试有旧数据 下面介绍两种清空数据的方法 一、通过…...
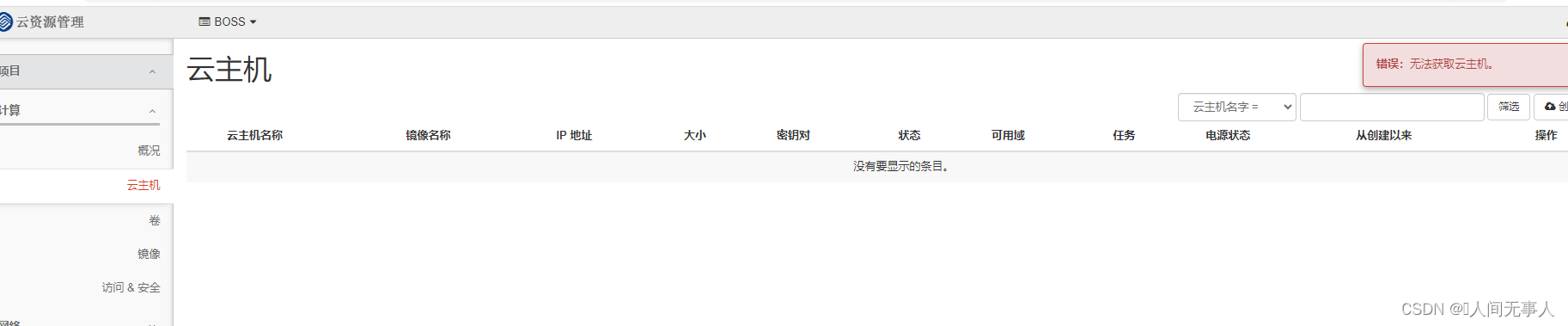
dashboard报错 错误:无法获取网络列表、dashboard报错 错误:无法获取云主机列表 解决流程
文章目录 错误说明dashboard上报错底层命令报错查看日志message日志httpd报错日志错误日志分析开始解决测试底层命令dashboard错误说明 dashboard上报错 首先,dashboard上无论是管理员还是其他项目,均无法获取云主机和网络信息,具体报错如下...

C语言中的3种注释方法
C语言中的3种注释方法 2021年8月28日星期六席锦 在用C语言编程时,常用的注释方式有如下几种: (1)单行注释 // … (2)多行注释 /* … */ (3)条件编译注释 #if 0…#endif (1)(2)在入门教程中比较常见。 对于(1) 【单行注释 // …】,注释只能显示…...
20款VS Code实用插件推荐
前言: VS Code是一个轻量级但功能强大的源代码编辑器,轻量级指的是下载下来的VS Code其实就是一个简单的编辑器,强大指的是支持多种语言的环境插件拓展,也正是因为这种支持插件式安装环境开发让VS Code成为了开发语言工具中的霸主…...

攻防世界web篇-robots
打开网址后,发现是一个空白页面的网页 但是,这个题目是robots,所以就联想到robots.txt这个目录,于是我就试了一下 注意:这里有个php的文件,这个应该就是一个目录文件 当输入后,直接回车&#…...

6 个可解锁部分 GPT-4 功能的 Chrome 扩展(无需支付 ChatGPT Plus 费用)
在过去的几个月里,我广泛探索了 ChatGPT 的所有可用插件。在此期间,我发现了一些令人惊叹的插件,它们改进了我使用 ChatGPT 的方式,但现在,我将透露一些您需要了解的内容。 借助 Chrome 扩展程序,所有 Chat…...

centos 7.9 安装sshpass
1.作用 sshpass是一个用于非交互式SSH密码验证的实用程序。它可以用于自动输入密码以进行SSH登录,从而简化了自动化脚本和批处理作业中的SSH连接过程。 sshpass命令可以与ssh命令一起使用,通过在命令行中提供密码参数来执行远程命令。以下是一个示例命…...

CompletableFuture多任务异步,获取返回值,汇总结果
线程池异步的基础知识 详情见:https://blog.csdn.net/sinat_32502451/article/details/133039624 线程池执行多任务,获取返回值 线程池的 submit()方法,可以提交任务,并返回 Future接口。 而 future.get(),可以获取…...
)
Linux上Qt和Opencv人脸识别项目学习路线(嵌入式/C++)
本文将介绍Linux人脸识别项目的开发流程, 只作简略介绍所需知识点及大致流程。 注:若需详细教程请联系作者(见文末)。 一、基本开发环境搭建 1.1 安装虚拟机Ubuntu 虚拟机采用的是VMware,需要下载VMware安装包、ubuntu系统镜像…...

spring 源码阅读之@Configuration解析
Configuration解析 Configuration注解用于标识一个类是配置类,用于声明和组织Bean定义,首先Configuration本身也是一个Component,在其注解定义上标有Component Target(ElementType.TYPE) Retention(RetentionPolicy.RUNTIME) Documented Co…...

MobileNetV3 医学病理分类:卷积分类头 + 迁移学习
文章目录 MobileNetV3 医学病理分类:卷积分类头 + 迁移学习 一、架构 二、环境 三、数据 3.1 结构 3.2 加载 四、模型 五、训练 六、推理 七、结果 八、卷积分类头 vs 全连接分类头 九、冻结 vs 微调对比 十、总结 代码链接与详细流程 购买即可解锁1000+YOLO优化文章,并且还有…...

Arm与RISC-V双架构OSM模块在工业控制中的应用
1. ARIES Embedded推出基于Renesas Arm/RISC-V的OSM模块在嵌入式系统领域,处理器架构的选择往往需要在Arm和RISC-V之间做出取舍。但ARIES Embedded最新发布的"MSRZG2UL"和"MSRZFive"系统级封装(SiP)模块打破了这一常规,同时提供了基…...

基于MCP协议构建技术术语翻译服务器:架构、集成与实战
1. 项目概述:一个为技术术语翻译而生的MCP服务器 如果你是一名开发者,尤其是在非英语母语环境下工作,或者你的项目需要面向多语言市场,那么你一定遇到过这样的场景:在阅读英文技术文档、编写代码注释,或者与…...

交通系统安全审计:90%的漏洞源于日志记录失误,你中招了吗?
🔥关注墨瑾轩,带你探索编程的奥秘!🚀 🔥超萌技术攻略,轻松晋级编程高手🚀 🔥技术宝库已备好,就等你来挖掘🚀 🔥订阅墨瑾轩,智趣学习不…...

自洽性与Agent的结合
让智能体学会“自我验证”,提升决策可靠性。随着大语言模型(LLM)从单纯的“对话接口”演进为“行动中枢”,AI Agent(智能体)正逐步突破“被动响应”的局限,向“自主决策、主动执行”的高阶形态演…...

玩转 Python:多线程、装饰器、视觉检测与正则匹配实战
Python 作为一门简洁又强大的编程语言,在多线程编程、函数增强、计算机视觉、文本处理等多个领域都有着广泛的应用。本文将结合几个实用的代码案例,带你上手 Python 的多线程、装饰器、OpenCV 颜色检测和正则表达式匹配,从基础应用到实际场景…...

刚开始做 GEO:最容易做错的动作与起步误区拆解
GEO 起步阶段,不建议先按“发多少内容、测多少平台、截多少图”做验收。 更合适的第一轮目标是:固定一批真实问题,检查公开材料能不能被 AI 正确组织成回答。讲不准,先修材料;讲得泛,先补边界;讲…...

simple_sq_music_plus
链接:https://pan.quark.cn/s/f4be936a9c8d预计更新时间不定 按照优先级排序酷狗概念喜欢自动下载(跟随3.0发布) docker-compose方便一键部署(跟随3.0发布))...
)
FreeModbus移植避坑指南:如何优雅地处理临界区与事件队列(含FreeRTOS示例)
FreeModbus在RTOS环境下的临界区与事件队列实战解析 当你第一次在FreeRTOS上成功运行FreeModbus时,那种成就感令人难忘。但很快,随着系统复杂度提升,随机崩溃、数据错乱、死锁等问题接踵而至——这几乎是每个嵌入式开发者都会经历的噩梦。不同…...

猫抓扩展:5分钟掌握网页视频下载与媒体提取的终极方案
猫抓扩展:5分钟掌握网页视频下载与媒体提取的终极方案 【免费下载链接】cat-catch 猫抓 浏览器资源嗅探扩展 / cat-catch Browser Resource Sniffing Extension 项目地址: https://gitcode.com/GitHub_Trending/ca/cat-catch 在互联网浏览中,你是…...