基于conda环境使用mamba/conda安装配置QIIME 2 2023.9 Amplicon扩增子分析环境,q2cli主要功能模块介绍及使用
QIIME 2 2023.9 Amplicon Distribution介绍:
概述
qiime团队专门针对高通量扩增子序列分析退出的conda集成环境,包括了主要和常见的扩增子分析模块,用户可以单独使用各个模块,也可以使用各模块组成不同的分析流程。从2023.09版本开始特意将扩增子和宏基因组shotgun分成了两个环境,可能一个环境集成太大了。这样安装应该更轻盈。
QIIME 2 2023.9 Amplicon Distribution
看看文章吧,超强的团队:Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 | Nature Biotechnology
2023.9Amplicon分发版qiime2主要功能模块介绍:
The 2023.9 release of the QIIME 2 Amplicon Distribution includes the QIIME 2 framework, q2cli (a QIIME 2 command-line interface) and the following plugins:
-
q2-alignment -
q2-composition -
q2-cutadapt -
q2-dada2 -
q2-deblur -
q2-demux -
q2-diversity -
q2-diversity-lib -
q2-emperor -
q2-feature-classifier -
q2-feature-table -
q2-fragment-insertion -
q2-longitudinal -
q2-metadata -
q2-phylogeny -
q2-quality-control -
q2-quality-filter -
q2-sample-classifier -
q2-taxa -
q2-types -
q2-vsearch
还有更多的模块,都可以单独安装,一般建议成套安装就行:
QIIME 2 Library

QIIME2的安装:
还是继续用conda吧,这个用起来也挺方便,docker稍微增加了点门槛,看个人喜好
官方安装介绍链接:
先看介绍选择分发板,根据自己喜好安装:Installing QIIME 2 — QIIME 2 2023.9.2 documentation

这里选择amplicon扩增子分析的环境:
查看安装指引:
Natively installing QIIME 2 — QIIME 2 2023.9.2 documentation
选择amplicon分发版的linux环境配置指导:

下载环境依赖文件并安装:
# 下载环境依赖配置文件
wget https://data.qiime2.org/distro/amplicon/qiime2-amplicon-2023.9-py38-linux-conda.yml# 使用mamba或conda 安装 ,-n后的名字自己定义就行
mamba env create -n qiime2-amplicon-2023.9 --file qiime2-amplicon-2023.9-py38-linux-conda.yml要升级qiime2?,建议按日期名称命名各个版本重新安装,不需要的就直接删除旧的环境,因为有些旧的环境下的模块大家可能还需要用到,而在新的环境下可能已经废弃或更新了,所以建议重新配置安装新的conda环境就行。
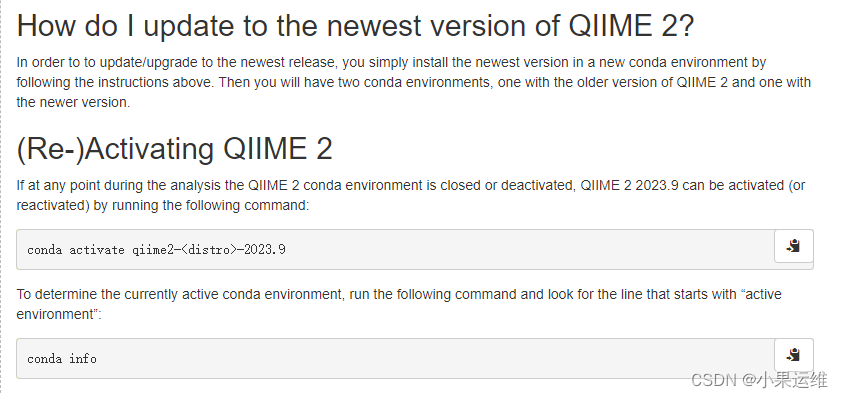
开始使用qiime2 amplicon分发版
要看怎么使用,或纠错,这里应该是最全的了,User Support - QIIME 2 Forum
这里来自刘永鑫团队的扩增子分析流程大家可以参考,把其中对应的步骤改为使用qiime模块分析即可:
使用vsearch进行16s扩增子高通量序列分析步骤-CSDN博客
EasyAmplicon (易扩增子)-扩增子高通量序列分析软件流程及脚本-详细使用方法——来自刘永鑫团队的秘籍-CSDN博客
当然qiime2 不仅仅提供分析模块,还开发了很多workflow,一个命令就可以按照workflow来得到结果了;可视化也有流程,后面大家看着参考:
Overview of QIIME 2 Plugin Workflows — QIIME 2 2023.9.2 documentation
先激活环境并查看环境信息
# 激活环境,conda或mamba
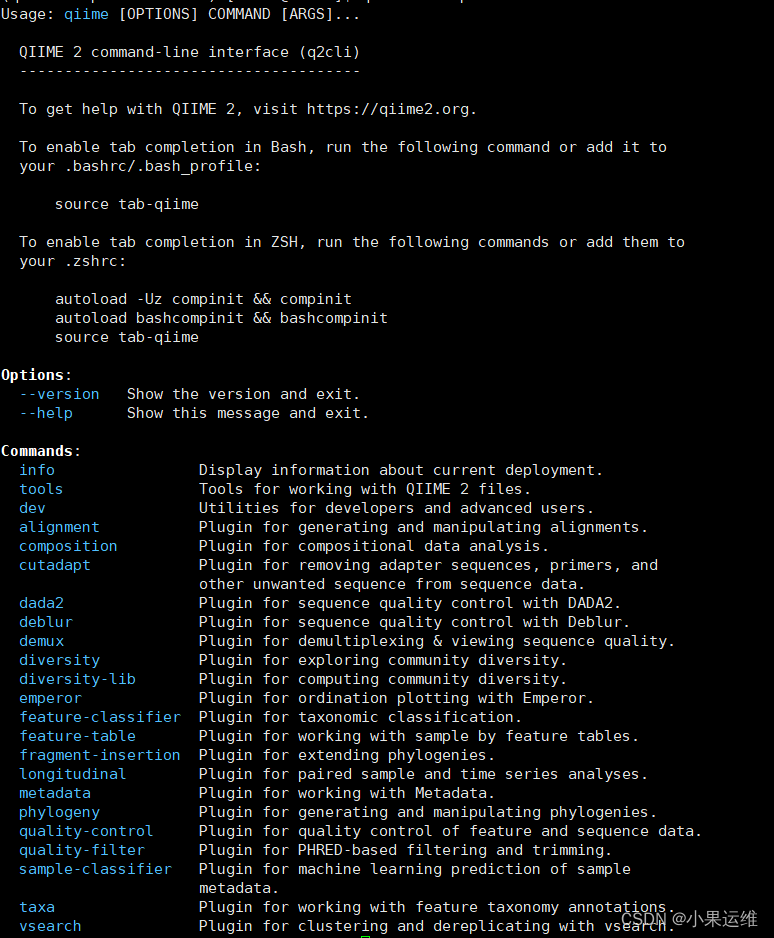
mamba activate qiime2-amplicon-2023.9# 查看帮助信息
qiime --help
开始qiime2分析
分析前建议大家先建立独立的工作目录,并进入工作目录再开始操作:
1、导入数据:
查看帮助:
![]()
qiime tools import --helpUsage: qiime tools import [OPTIONS]Import data to create a new QIIME 2 Artifact. See https://docs.qiime2.org/for usage examples and details on the file types and associated semantictypes that can be imported.Options:--type TEXT The semantic type of the artifact that will becreated upon importing. Use --show-importable-typesto see what importable semantic types are availablein the current deployment. [required]--input-path PATH Path to file or directory that should be imported.[required]--output-path ARTIFACT Path where output artifact should be written.[required]--input-format TEXT The format of the data to be imported. If notprovided, data must be in the format expected by thesemantic type provided via --type.--show-importable-types Show the semantic types that can be supplied to--type to import data into an artifact.--show-importable-formatsShow formats that can be supplied to --input-formatto import data into an artifact.--help Show this message and exit.
大家可以先使用样例文件做为参考,然后按样例文件整理自己的数据
双端序列导入(混合样品测序,序列文件带barcode,分析时需要导入样品的metadata)Multiplexed paired-end FASTQ with barcodes in sequence
这种应该符合大家现在常规混合样品测序的情况了。
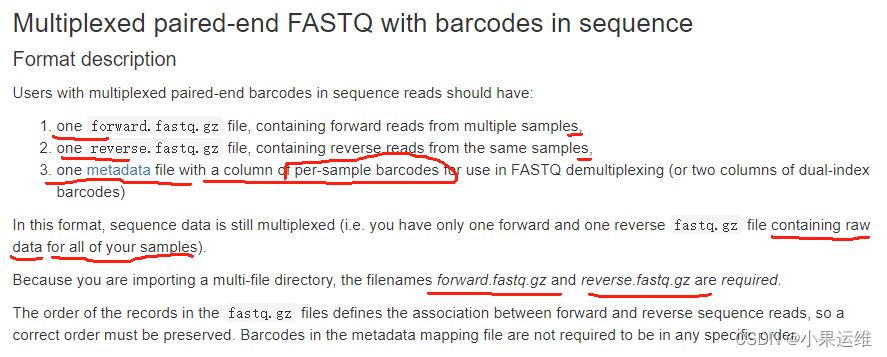
metadata格式,重要的是第一列和第二列,或者再加一列barcode2
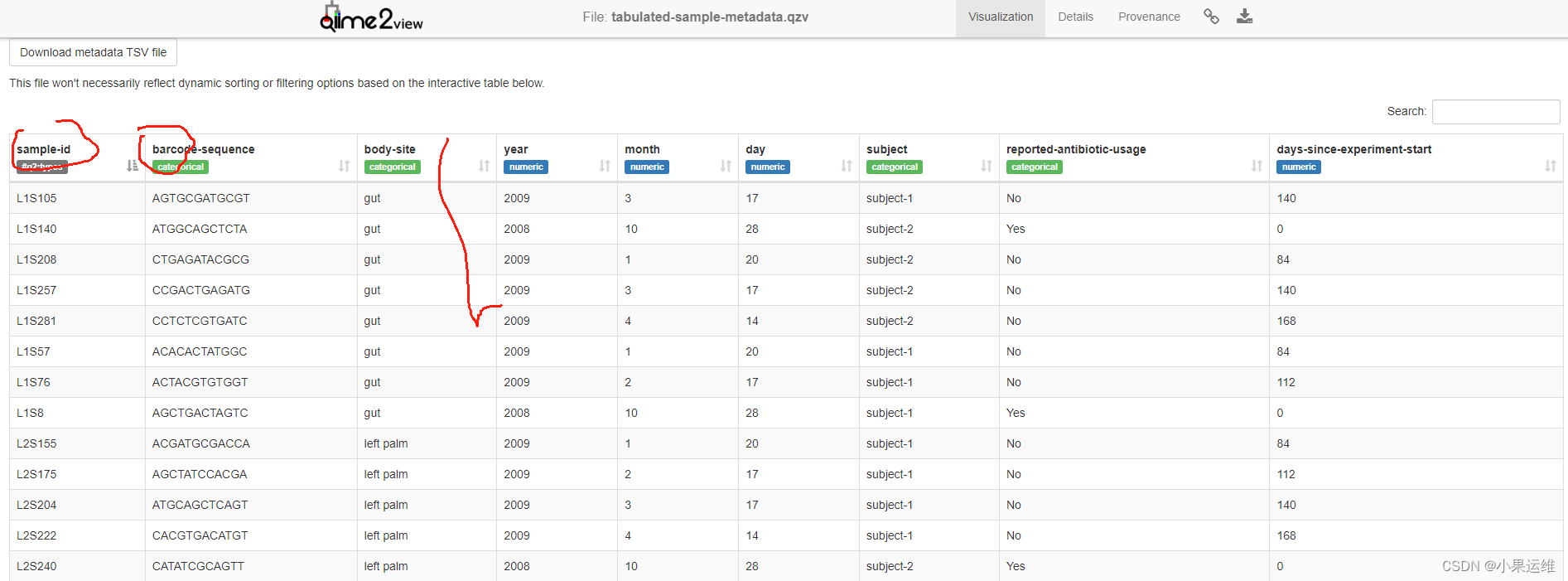
序列文件导入(三个文件,forward.fastq.gz, reverse.fastq.gz, sample_metadata.tsv):
qiime tools import \--type MultiplexedPairedEndBarcodeInSequence \--input-path muxed-pe-barcode-in-seq \--output-path multiplexed-seqs.qza单端序列导入(序列文件与barcode文件各自独立)
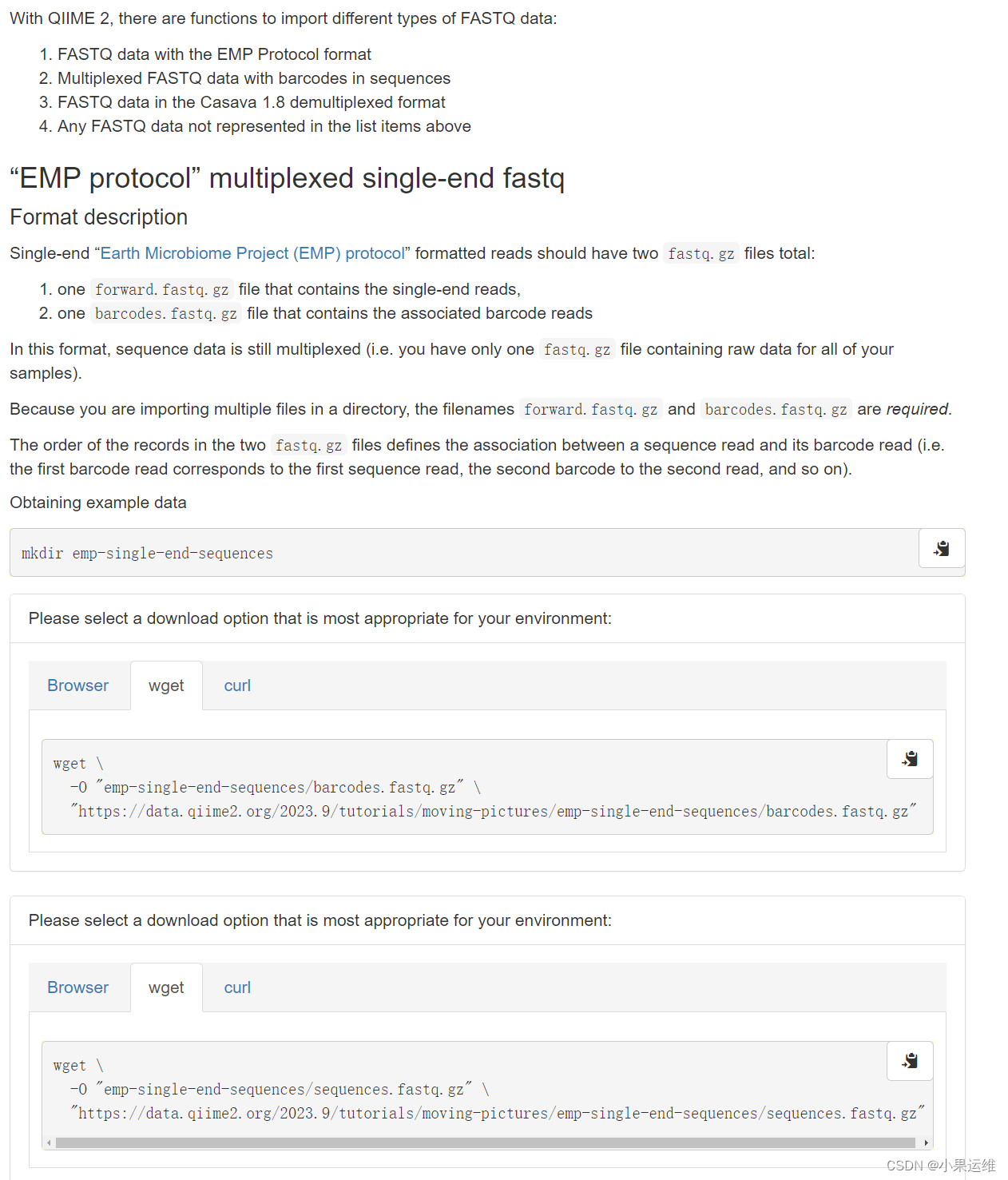
复制官方代码时注意删除上面的引号,这个复制过来容易变成中文的双引号,另外还需要注意存储路径的设置 :

获取样例文件
# 进入工作目录
cd emp-single-end-sequences/# barcode文件
wget \-O barcodes.fastq.gz \https://data.qiime2.org/2023.9/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz# 序列文件,这里是单端序列
wget \-O sequences.fastq.gz \https://data.qiime2.org/2023.9/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz
查看要导入的文件格式:
#
zcat barcodes.fastq.gz | head -n 10
# barcode文件内容,
@HWI-EAS440_0386:1:23:17547:1423#0/1
ATGCAGCTCAGT
+
IIIIIIIIIIIH
@HWI-EAS440_0386:1:23:14818:1533#0/1
CCCCTCAGCGGC
+
DDD@D?@B<<+/
@HWI-EAS440_0386:1:23:14401:1629#0/1
GACGAGTCAGTC#
zcat sequences.fastq.gz | head -n 10# sequences文件内容
@HWI-EAS440_0386:1:23:17547:1423#0/1
TACGNAGGATCCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGATGGATGTTTAAGTCAGTTGTGAAAGTTTGCGGCTCAACCGTAAAATTGCAGTTGATACTGGATATCTTGAGTGCAGTTGAGGCAGGGGGGGATTGGTGTG
+
IIIE)EEEEEEEEGFIIGIIIHIHHGIIIGIIHHHGIIHGHEGDGIFIGEHGIHHGHHGHHGGHEEGHEGGEHEBBHBBEEDCEDDD>B?BE@@B>@@@@@CB@ABA@@?@@=>?08;3=;==8:5;@6?######################
@HWI-EAS440_0386:1:23:14818:1533#0/1
CCCCNCAGCGGCAAAAATTAAAATTTTTACCGCTTCGGCGTTATAGCCTCACACTCAATCTTTTATCACGAAGTCATGATTGAATCGCGAGTGGTCGGCAGATTGCGATAAACGGGCACATTAAATTTAAACTGATGATTCCACTGCAACAA
+
64<2$24;1)/:*B<?BBDDBBD<>BDD############################################################################################################################
@HWI-EAS440_0386:1:23:14401:1629#0/1
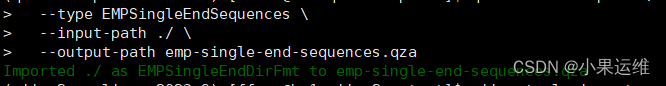
TACGNAGGATCCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGGCGGACGCTTAAGTCAGTTGTGAAAGTTTGCGGCTCAACCGTAAAATTGCAGTTGATACTGGGTGTCTTGAGTACAGTAGAGGCAGGGGGGGGGTTGGGGG## 激活 qiime2环境:## 导入数据
qiime tools import \--type EMPSingleEndSequences \--input-path ./ \--output-path emp-single-end-sequences.qza#### qiime tools import --show-importable-types 这个命令已经废弃了,大家注意在新版里没法用qiime tools list-types
Usage: qiime tools list-types [OPTIONS] [QUERIES]...List the available semantic types.Options:--strict Show only exact matches for the type argument(s).--tsv Print as machine readable tab-separated values.--help Show this message and exit.
双端序列导入(序列文件与barcode文件各自独立)

获取样例文件
cd emp-paired-end-sequences
# 下载正向序列
wget \-O forward.fastq.gz \https://data.qiime2.org/2023.9/tutorials/atacama-soils/1p/forward.fastq.gz#下载反向序列
wget \-O reverse.fastq.gz \https://data.qiime2.org/2023.9/tutorials/atacama-soils/1p/reverse.fastq.gz# 下载barcodes文件
wget \-O barcodes.fastq.gz \https://data.qiime2.org/2023.9/tutorials/atacama-soils/1p/barcodes.fastq.gz# 查看输入文件格式
# 正向序列文件内容
zcat forward.fastq.gz | head -n 10
@M00176:65:000000000-A41FR:1:1101:14282:1412 1:N:0:0
NACGTAGGGTGCAAGCGTTAATCGGAATTACNGGNNNTAAAGCGTGCNNAGGCNNNNNNNNNNNANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
#>>>AAF@ACAA4BGCEEECGGHGGEFCFBG#BA###BABAEFGEEE##BBAA###########B######################################################################################
@M00176:65:000000000-A41FR:1:1101:16939:1420 1:N:0:0
NACGTAGGGGGCAAGCGTTGTCCGGAATCATTGGNNGTAAAGAGCGTGNAGGCNNNNNGNNANNTNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
+
#>>ABAF@ABBBGGGGEEFGGGHGGFFGFHHHHH##BBEFDHHCGGFG#ABFF#####B##B##B######################################################################################
@M00176:65:000000000-A41FR:1:1101:14746:1560 1:N:0:0
TACGTAGGGAGCTAGCGTTGTCCGGAATCATTGGGCGTAAAGCGCGCGTAGGCGGCCAGATAAGTCCGGTGTAAAAGCCACAGGCTNNNNNNNNNNNNNNNNCNGGANNNNNNNNNNNNNNNNNNNNNNANNNNNNNNNNNNANNNNNGGN# 反向序列文件内容
zcat reverse.fastq.gz | head -n 10
@M00176:65:000000000-A41FR:1:1101:14282:1412 2:N:0:0
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCGGAATNCCATNCCNCTCTGCNNNNNNNNNNNNNNNNNNNNNNNNN
+
#########################################################################################################<</?F/#/</?#?<#<???/<#########################
@M00176:65:000000000-A41FR:1:1101:16939:1420 2:N:0:0
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCNCNNTNCNNNNNNNNNNNGGGAATTCCACNCGCCTCTTCNNNNNNNNNNNNNNNNNNNNNNNNN
+
######################################################################################?#?##?#?###########<??/?FC>FBF#</??FAGHG#########################
@M00176:65:000000000-A41FR:1:1101:14746:1560 2:N:0:0
NNNNTTTCGTNGATNNNNNNNCNGNNNNNNNNNNNNNNNNNNNTNNNNNNNTACCNNNNNNNNTGGGCCTCTGTGCGCATACCGTTCGAGAGCGCCGGCCACGGCATTCGTGTTGCTCCTCTAAGCTGTGCGTGTTCACTGTACAACCNNN# barcode文件内容
zcat barcodes.fastq.gz | head -n 10
@M00176:65:000000000-A41FR:1:1101:14282:1412 1:N:0:0
NNNNNNNNNNNN
+
############
@M00176:65:000000000-A41FR:1:1101:16939:1420 1:N:0:0
NNNNNNNNNNNN
+
############
@M00176:65:000000000-A41FR:1:1101:14746:1560 1:N:0:0
NNNNNNNNNNNN
数据导入:
cd emp-paired-end-sequencesqiime tools import \--type EMPPairedEndSequences \--input-path ./ \--output-path emp-paired-end-sequences.qza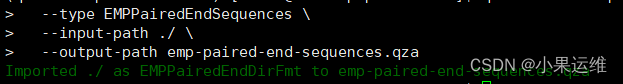
带barcodes的单端序列文件导入

获取样例文件
#
cd muxed-se-barcode-in-seq/
wget \-O sequences.fastq.gz \https://data.qiime2.org/2023.9/tutorials/importing/muxed-se-barcode-in-seq.fastq.gz# 查看输入文件格式:
zcat sequences.fastq.gz | head -n 20
@M00899:113:000000000-A5K20:1:1101:18850:2539 1:N:0:2
GCTACGGGGGGCAGCAGTGGGGAATATTGCACAATGGGCGAAAGCCTGATGCAGCAACGCCGCGTGAACGATGAAGGTCTTCGGATCGTAAAGTTCTGTTGCAGGGGAAGATAATGACGGTACCCTGTGAGGAAGCCCCGGCTAACTACGTGCCAGCAGCCGCGGTAATACGTAGGGGGCTAGCGTTATCCGGATTTACTGGGCGTAAAGGGTGCGTAGGTGGTCCTTCAAGTCGGTGGTTAAAGGCTAAGGCTCAACCGTAGTAAGCCGCCGAAACTGGAGGACTTGAGTGAAGGAGAGG
+
-8ABCC>=>5811884:<:99=?@EECFFGDFADECFFFEEDDEFEDEDFFFEEFCCBCF>CCB3CFF:BBFFFCCD,8@9C@C:+5@@:A@C<FDCFBEG>FFFDGCCEC?FGGGGGGGGGCFGGFCFGGGGGGGGGGEG7CFFGFFFGGGFG?FACE;:8CCCCEEF9<F@FFEGGC**/:3:2CC@:C;C81;C9<?>FF8C758CGGG2:7DC>EECEFE9+27CF492/8B7>D)7@F=FFCFF*9F52<2,289<0:44AB<49(3<>F51).69D?D34*44:4<5<B?::086
@M00899:113:000000000-A5K20:1:1101:25454:3578 1:N:0:2
CCTACGGGAGGCAGCAGTGAGGAATATTGGTCAATGGGCGAGAGCCTGAACCAGCCAAGTAGCGTGCAGGATGACGGCCCTATGGGTTGTAAACTGCTTTTGTATGGGGATAAAGTCAGTCACGTGTGATTGTTTGCAGGTACCATACGAATAAGGACCGGCTAATTCCGTGCCAGCAGCCGCGGTAATACGGAAGGTCCGGGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGGCTGGAGATTAAGTGTGTTGTGAAATGTAGACGCTCAACGTCTGACTTGCAGCGCATACTGG
+
8ACCCGD@AA=18=======;CEFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGFFF?FGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCGFGGGFFFGGGGGGEFGGGGGGGGGGGGCGEGGDGGGGGGGGGG=CGGCECDGGFGGGGGGGGFGGGF>C>BFFEGFFFFF:FGGF=6=6:AFBGFFFFFFA9A<AFB?@0)>C:0<CF?C46FAD<??90;::?DA>
@M00899:113:000000000-A5K20:1:1101:25177:3605 1:N:0:2
CCTACGGGAGGCAGCAGTGAGGAATATTGGTCAATGGACGGAAGTCTGAACCAGCCAAGTAGCGTGCAGGATGACGGCCCTATGGGTTGTAAACTGCTTTTGTATGGGGATAAAGTTAGGGACGTGTCCCTATTTGCAGGTACCATACGAATAAGGACCGGCTAATTCCGTGCCAGCAGCCGCGGTAATACGGAAGGTCCAGGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGGCTGGAGATTAAGTGTGTTGTGAAATGTAGACGCTCAACGTCTGAATTGCAGCGCATACTGG
+
88BCCEDAD9018======;;CCFGGGGFGGGFGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGEGDGGGGGGGGGFFGGGGGGGGGFGGGFGGGFGGGFFGCGGGGGGFGGFDGGGGGGGGGGGGG5CBFGCGGGGC?FGGGGGGGGGGGDEGDDDGFGGGGGEGGGGGGA39>BFFDDEF4:D5@CE?CFFF>>ABGFFF9<A246<<<<B::DE=<?FGGFG4>F?DF?02211:DAF7### barcode已经在序列中导入文件:
qiime tools import \--type MultiplexedSingleEndBarcodeInSequence \--input-path sequences.fastq.gz \--output-path multiplexed-seqs.qza
当然还有其他可导入文件格式,慢慢看吧,个人觉得有需要的时候查一下就行,不用全部了解:
qiime tools list-formats --importable2、导出数据
官方示例,大家可以下载下来测试一下就行了,不再细讲了。
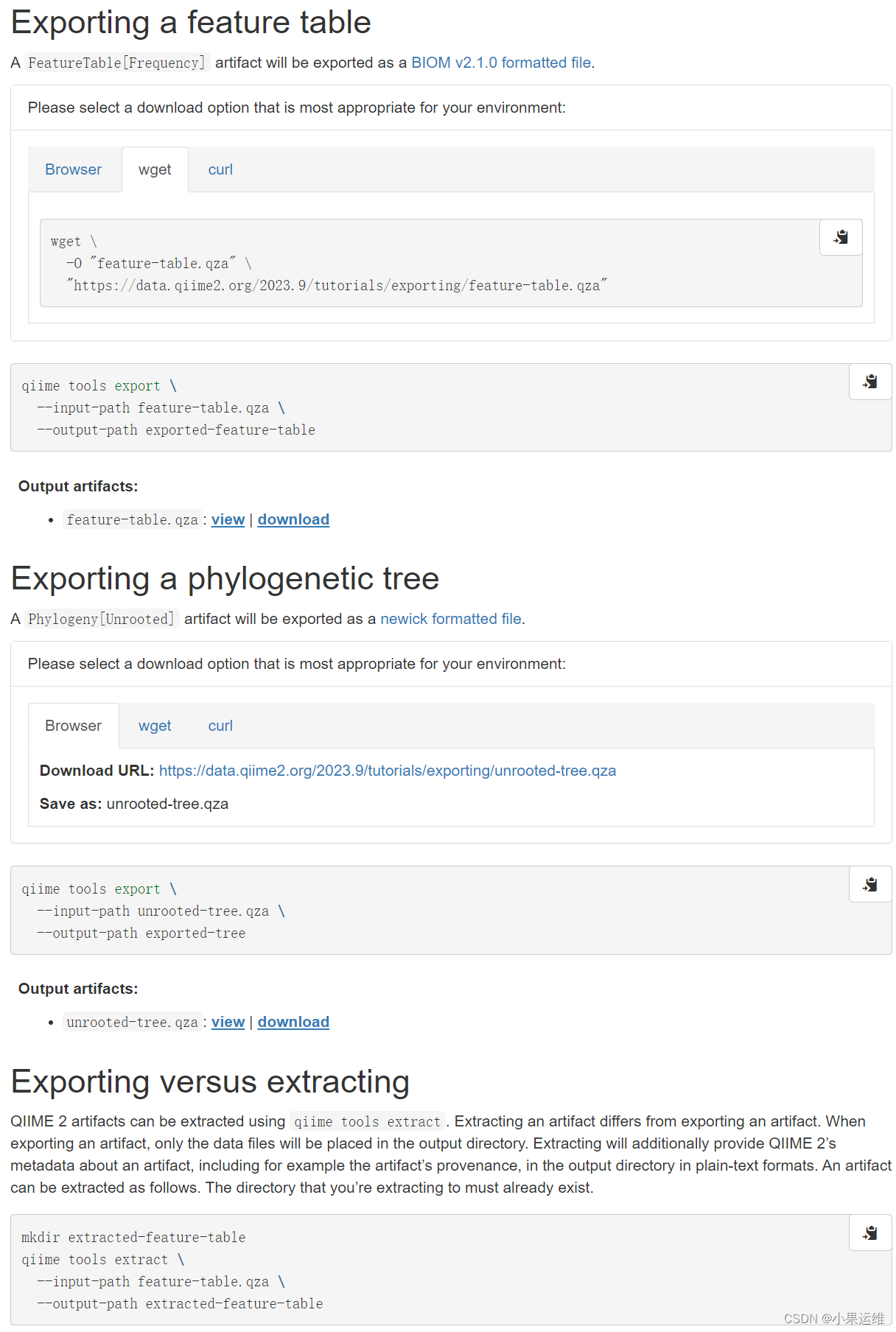
下载测试:
wget \-O "feature-table.qza" \"https://data.qiime2.org/2023.9/tutorials/exporting/feature-table.qza"wget \-O "unrooted-tree.qza" \"https://data.qiime2.org/2023.9/tutorials/exporting/unrooted-tree.qza"qiime tools export \--input-path feature-table.qza \--output-path exported-feature-table## 导出为biom格式文件
## 参考:https://biom-format.org/documentation/format_versions/biom-2.1.htmlqiime tools export \--input-path unrooted-tree.qza \--output-path exported-tree## 导出为.nwk文件,可在各类软件再直接出发育树的图
head tree.nwk
(((New.CleanUp.ReferenceOTU1480:0.11995,(New.CleanUp.ReferenceOTU202:0.04479,New.CleanUp.ReferenceOTU432:0.0049)0.769:0.04661)1:0.26705,((New.CleanUp.ReferenceOTU1150:0.00016,(New.CleanUp.ReferenceOTU782:0.04264,(New.CleanUp.ReferenceOTU643:0.10438,(((New.CleanUp.ReferenceOTU1014:0.01521,New.CleanUp.ReferenceOTU270:0.02738)0.879:0.02315,(((New.CleanUp.ReferenceOTU1008:0.0378,(New.CleanUp.ReferenceOTU1222:0.01621,(New.CleanUp.ReferenceOTU230:0.01829,(New.CleanUp.ReferenceOTU1047:0.0303,New.CleanUp.ReferenceOTU605:0.00015)0.88:0.01596)0.871:0.01515)0.714:0.01214)0.996:0.06757,(New.CleanUp.ReferenceOTU681:0.00441,(New.CleanUp.ReferenceOTU1485:0.01177,New.CleanUp.ReferenceOTU130:0.05699)0.923:0.02883)0.367:0.02397)0.17:0.00665,(New.CleanUp.ReferenceOTU1330:0.0258,(New.CleanUp.ReferenceOTU903:0.04234,(((New.CleanUp.ReferenceOTU1077:0.00014,(New.CleanUp.ReferenceOTU582:0.0171,New.CleanUp.ReferenceOTU987:0.0226)0.951:0.02372)0.892:0.02206,(New.CleanUp.ReferenceOTU891:0.01038,(((New.CleanUp.ReferenceOTU1066:0.01299,New.CleanUp.ReferenceOTU764:0.02254)0.851:0.01818,(New.CleanUp.ReferenceOTU180:0.02021,New.CleanUp.ReferenceOTU535:0.00014)0.795:0.01084)0.921:0.02441,(New.CleanUp.ReferenceOTU1212:0.00521mkdir extracted-feature-table
qiime tools extract \--input-path feature-table.qza \--output-path extracted-feature-table## 3、样品元数据处理
这个可能得科学上网,给大家下载了,大家参考下载吧。
https://download.csdn.net/download/zrc_xiaoguo/88616825?spm=1001.2014.3001.5503
样例文件列表:
sample-metadata数据:

元数据表qiime2转换命令:
qiime metadata tabulate \--m-input-file sample-metadata.tsv \--o-visualization tabulated-sample-metadata.qzvqiime metadata tabulate \--m-input-file faith_pd_vector.qza \--o-visualization tabulated-faith-pd-metadata.qzvqiime metadata tabulate \--m-input-file sample-metadata.tsv \--m-input-file faith_pd_vector.qza \--o-visualization tabulated-combined-metadata.qzvqiime emperor plot \--i-pcoa unweighted_unifrac_pcoa_results.qza \--m-metadata-file sample-metadata.tsv \--m-metadata-file faith_pd_vector.qza \--o-visualization unweighted-unifrac-emperor-with-alpha.qzv转换后数据qzv查看
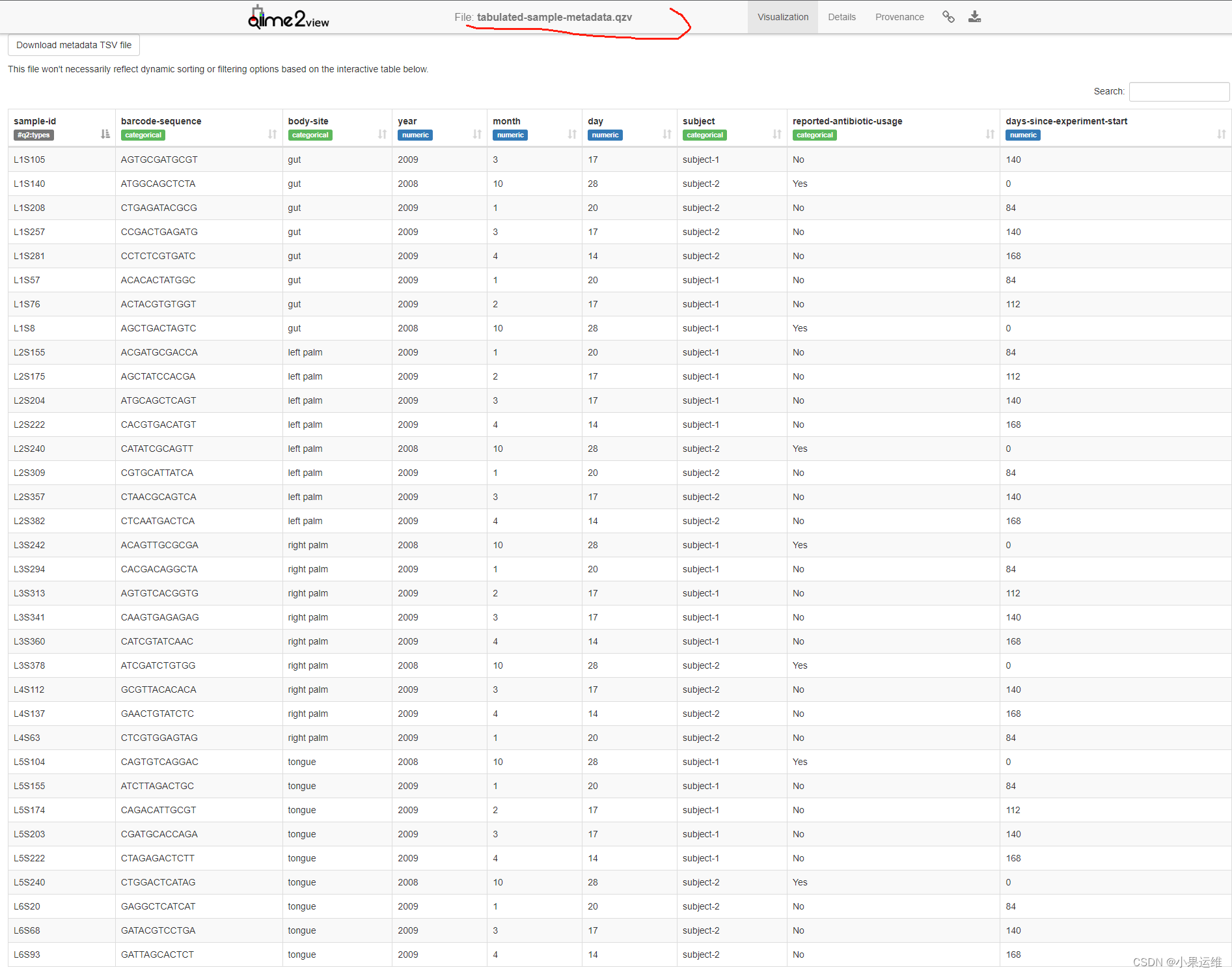
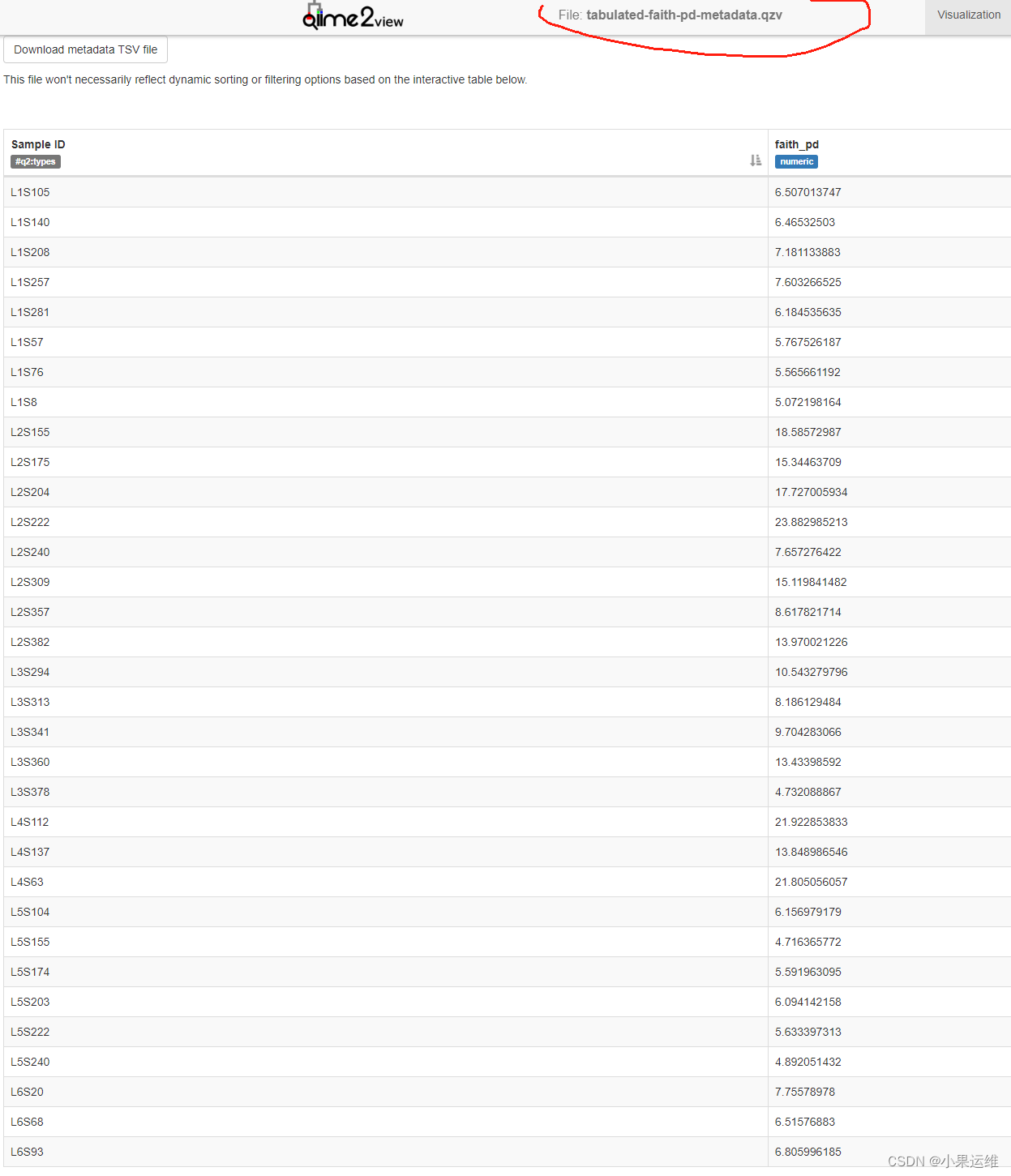
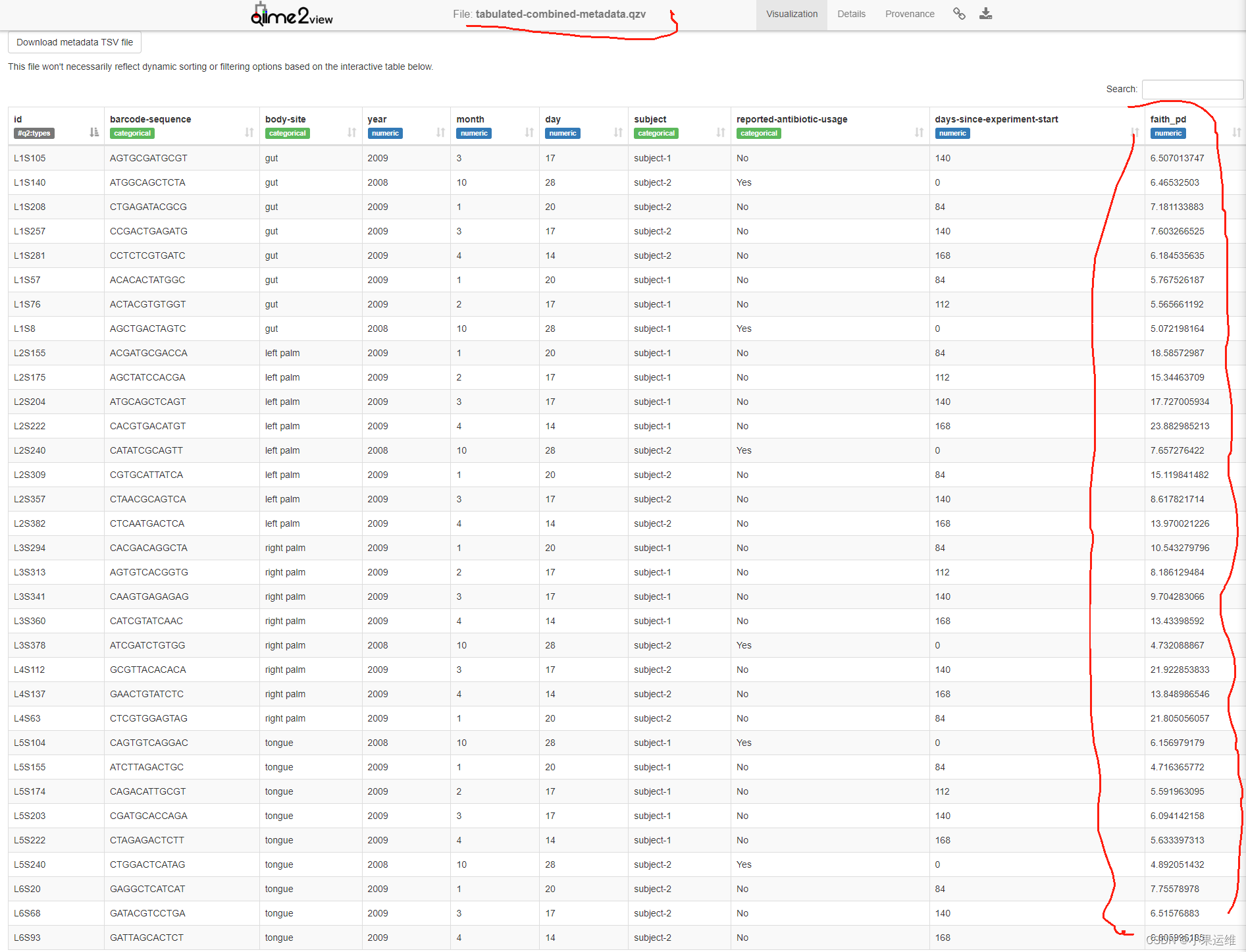
4、数据过滤
Filtering data — QIIME 2 2023.9.2 documentation
特征表过滤Filtering feature tables
基于总出现频率过滤,去掉低丰度序列,或筛选一定丰度的序列,比如说将频率低于1500的序列去掉,包括两个参数,最高和最低频率
The --p-min-frequency and --p-max-frequency can be combined to filter based on lower and upper limits of total frequency
这里只给了一个最低频率的限制。
qiime feature-table filter-samples \--i-table table.qza \--p-min-frequency 1500 \--o-filtered-table sample-frequency-filtered-table.qza基于出现的偶然性的过滤???Contingency-based filtering,也就是最少在多少个样品中都需要出现,通量有最高和最低频率设定: --p-min-features and --p-min-samples。
qiime feature-table filter-features \--i-table table.qza \--p-min-samples 2 \--o-filtered-table sample-contingency-filtered-table.qza字段筛选Identifier-based filtering, 也就是保留哪些特征数据内容。
qiime feature-table filter-samples \--i-table table.qza \--m-metadata-file samples-to-keep.tsv \--o-filtered-table id-filtered-table.qza同样还有很多其他的筛选方式:
Metadata-based filtering
Taxonomy-based filtering of tables and sequences
序列过滤Filtering sequences
比如说这里根据原序列文件和分类文件提取mitochondria,chloroplast这两个门水平的序列文件。
qiime taxa filter-seqs \--i-sequences sequences.qza \--i-taxonomy taxonomy.qza \--p-include p__ \--p-exclude mitochondria,chloroplast \--o-filtered-sequences sequences-with-phyla-no-mitochondria-no-chloroplast.qzaFiltering distance matrices
qiime diversity filter-distance-matrix \--i-distance-matrix distance-matrix.qza \--m-metadata-file samples-to-keep.tsv \--o-filtered-distance-matrix identifier-filtered-distance-matrix.qzaqiime diversity filter-distance-matrix \--i-distance-matrix distance-matrix.qza \--m-metadata-file sample-metadata.tsv \--p-where "[subject]='subject-2'" \--o-filtered-distance-matrix subject-2-filtered-distance-matrix.qza5、重要参考数据库
这个不多说了吧,大家按使用需求来下载,后面逐步会用到:
Data resources — QIIME 2 2023.9.2 documentation
6、各个插件模块使用
不熟悉的先查看help信息和官网介绍:
-
q2-alignment
q2-alignment 是 QIIME 2 生态系统中的一个插件,用于对生物信息学序列数据进行比对和序列比较的工具。它可以用于将不同样本中的序列进行比对,分析它们之间的相似性和差异性。q2-alignment 提供了一些常用的比对算法和工具,使用户能够对序列进行比对、生成比对结果和后续的分析。
以下是 q2-alignment 插件的一些主要功能和使用方法:
功能和用途:
- 序列比对:对DNA、RNA或蛋白质序列进行比对。
- 多序列比对:能够处理多个序列文件,比对它们之间的相似性。
- 生成比对结果:产生比对结果,以便后续分析或可视化。
- 支持多种比对算法:包括常用的比对算法,如BLAST、MUSCLE、MAFFT 等。
qiime alignment --help
Usage: qiime alignment [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin provides support for generating andmanipulating sequence alignments.Plugin website: https://github.com/qiime2/q2-alignmentGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:mafft De novo multiple sequence alignment with MAFFTmafft-add Add sequences to multiple sequence alignment with MAFFT.mask Positional conservation and gap filtering.
# 运行比对(示例使用 MAFFT)
qiime alignment mafft \--i-sequences sequences.qza \--o-alignment aligned_sequences.qza-
q2-composition
用于进行组成分析,尤其是在处理微生物组数据中的相对丰度数据时非常有用。该插件可以帮助用户探索和比较微生物组中微生物群落的组成。
qiime composition --help
Usage: qiime composition [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports methods for compositional dataanalysis.Plugin website: https://github.com/qiime2/q2-compositionGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:add-pseudocount Add pseudocount to table.ancom Apply ANCOM to identify features that differ in abundance.ancombc Analysis of Composition of Microbiomes with Bias Correctionda-barplot Differential abundance bar plotstabulate View tabular output from ANCOM-BC.
# 导入数据
qiime composition add-pseudocount \--i-table table.qza \--o-composition-table composition.qza#数据转换
qiime composition ilr-transform \--i-table composition.qza \--o-transformed-table ilr_composition.qza#可视化分析结果
qiime composition pcoa \--i-table ilr_composition.qza \--o-pcoa ilr_composition_pcoa.qzaqiime emperor plot \--i-pcoa ilr_composition_pcoa.qza \--m-metadata-file metadata.txt \--o-visualization ilr_composition_emperor.qzv
q2-cutadapt
q2-cutadapt插件是用于对DNA序列数据进行预处理和过滤的工具。它基于Cutadapt软件,允许用户对Illumina测序数据进行裁剪(trimming)、过滤(filtering)和修剪(adapter removal)等操作,以消除低质量序列、去除适配器、修剪序列末端等。这有助于提高序列数据的质量,为后续的分析准备干净、高质量的数据。
#
qiime cutadapt --help
Usage: qiime cutadapt [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin uses cutadapt to work with adapters (e.g.barcodes, primers) in sequence data.Plugin website: https://github.com/qiime2/q2-cutadaptGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:demux-paired Demultiplex paired-end sequence data with barcodes in-sequence.demux-single Demultiplex single-end sequence data with barcodes in-sequence.trim-paired Find and remove adapters in demultiplexed paired-endsequences.trim-single Find and remove adapters in demultiplexed single-endsequences.qiime cutadapt trim-paired \--i-demultiplexed-sequences demux.qza \--p-cores 8 \--p-front-f CCTACGGGNGGCWGCAG \--p-front-r GACTACHVGGGTATCTAATCC \--p-discard-untrimmed \--o-trimmed-sequences demux_trimmed.qza参数解释:
--i-demultiplexed-sequences: 输入的序列文件(需提前导入到QIIME 2)。
--p-cores: 并行处理的CPU核心数量。
--p-front-f 和 --p-front-r: 正向和反向引物序列。
--p-discard-untrimmed: 丢弃未被修剪的序列。
--o-trimmed-sequences: 输出修剪后的序列文件。
-
q2-dada2
q2-dada2是QIIME 2的插件之一,基于DADA2算法,用于去噪和分析Illumina测序生成的16S rRNA数据。
qiime dada2 --help
Usage: qiime dada2 [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin wraps DADA2 and supports sequence qualitycontrol for single-end and paired-end reads using the DADA2 R library.Plugin website: http://benjjneb.github.io/dada2/Getting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:denoise-ccs Denoise and dereplicate single-end Pacbio CCSdenoise-paired Denoise and dereplicate paired-end sequencesdenoise-pyro Denoise and dereplicate single-end pyrosequencesdenoise-single Denoise and dereplicate single-end sequences
qiime dada2 denoise-single \--i-demultiplexed-seqs input-demux.qza \--p-trim-left 0 \--p-trunc-len 120 \--o-representative-sequences rep-seqs-dada2.qza \--o-table table-dada2.qza \--o-denoising-stats stats-dada2.qza参数说明:
--i-demultiplexed-seqs:输入的demultiplexed序列文件。--p-trim-left:要去除的序列的前部分碱基数量。--p-trunc-len:截断序列的长度。--o-representative-sequences:输出的代表序列文件。--o-table:生成的特征表文件。--o-denoising-stats:生成的去噪统计文件。
-
q2-deblur
q2-deblur插件通过识别和去除16S rRNA基因测序数据中的测序错误和噪声,以生成高质量的序列数据。其主要步骤包括:
- 生成特征表(Feature table): 从原始的FASTQ格式文件中导入数据,创建特征表。
- 质量过滤: 过滤低质量序列,去除低质量序列读数。
- 去噪处理: 使用Deblur算法去除测序错误和噪声,生成高质量的特征序列。
- 生成结果: 输出一个经过去噪处理和质量过滤的特征表和序列文件。
qiime deblur --help
Usage: qiime deblur [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin wraps the Deblur software for performingsequence quality control.Plugin website: https://github.com/biocore/deblurGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:denoise-16S Deblur sequences using a 16S positive filter.denoise-other Deblur sequences using a user-specified positive filter.visualize-stats Visualize Deblur stats per sample.
qiime deblur denoise-16S \--i-demultiplexed-seqs your_qza_file.qza \--p-trim-length 250 \ # 设置序列截断长度--o-representative-sequences rep_seqs.qza \--o-table table.qza \--p-sample-stats \ # 如果需要生成样本统计信息--o-stats deblur_stats.qza
结果数据导出
# 结果数据导出
qiime tools export --input-path rep_seqs.qza --output-path exported_rep_seqs
qiime tools export --input-path table.qza --output-path exported_table
qiime tools export --input-path deblur_stats.qza --output-path exported_stats
-
q2-demux
q2-demux 插件用于处理 DNA 或 RNA 测序数据的样本数据解复用(demultiplexing)和质量控制。这个插件允许用户根据样本的不同 DNA 或 RNA 序列标签(barcode 或者 Illumina 测序的 index)将混合测序数据集拆分成单独的样本。下面是 q2-demux 插件的基本介绍和使用步骤:
q2-demux 插件功能:
-
数据解复用(Demultiplexing):
- 将混合测序数据根据每个样本的唯一标识(barcode 或 index)分割成单独的样本序列文件。
-
质量控制:
- 提供了检查序列数据质量的功能,允许用户查看样本数据的质量分数并进行必要的处理,比如修剪或过滤低质量序列。
qiime demux --help
Usage: qiime demux [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports demultiplexing of single-end andpaired-end sequence reads and visualization of sequence quality information.Plugin website: https://github.com/qiime2/q2-demuxGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:emp-paired Demultiplex paired-end sequence data generatedwith the EMP protocol.emp-single Demultiplex sequence data generated with the EMPprotocol.filter-samples Filter samples out of demultiplexed data.partition-samples-paired Split demultiplexed sequence data into partitions.partition-samples-single Split demultiplexed sequence data into partitions.subsample-paired Subsample paired-end sequences withoutreplacement.subsample-single Subsample single-end sequences withoutreplacement.summarize Summarize counts per sample.tabulate-read-counts Tabulate counts per sample
# 执行去重复
qiime demux emp-paired \--i-seqs demux.qza \--m-barcodes-file sample-metadata.tsv \--m-barcodes-column BarcodeSequence \--o-per-sample-sequences demux-paired-end.qza \--o-error-correction-details demux-details.qza## 查看结果
qiime demux summarize \--i-data demux-paired-end.qza \--o-visualization demux-summary.qzv
-
q2-diversity
q2-diversity是其中一个用于计算和分析生物多样性的插件。它可以帮助你评估样本群落的多样性和差异性。
qiime diversity --help
Usage: qiime diversity [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports metrics for calculating andexploring community alpha and beta diversity through statistics andvisualizations in the context of sample metadata.Plugin website: https://github.com/qiime2/q2-diversityGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:adonis adonis PERMANOVA test for beta group significancealpha Alpha diversityalpha-correlation Alpha diversity correlationalpha-group-significance Alpha diversity comparisonsalpha-phylogenetic Alpha diversity (phylogenetic)alpha-rarefaction Alpha rarefaction curvesbeta Beta diversitybeta-correlation Beta diversity correlationbeta-group-significance Beta diversity group significancebeta-phylogenetic Beta diversity (phylogenetic)beta-rarefaction Beta diversity rarefactionbioenv bioenvcore-metrics Core diversity metrics (non-phylogenetic)core-metrics-phylogenetic Core diversity metrics (phylogenetic and non-phylogenetic)filter-distance-matrix Filter samples from a distance matrix.mantel Apply the Mantel test to two distance matricespartial-procrustes Partial Procrustespcoa Principal Coordinate Analysispcoa-biplot Principal Coordinate Analysis Biplotprocrustes-analysis Procrustes Analysistsne t-distributed stochastic neighbor embeddingumap Uniform Manifold Approximation and Projection
# Alpha多样性(样本内部多样性)
# 计算Shannon指数:
qiime diversity alpha \--i-table your_feature_table.qza \--p-metric shannon \--o-alpha-diversity shannon_vector.qza#Beta多样性(样本间多样性)
# 计算Bray-Curtis距离:qiime diversity beta \--i-table your_feature_table.qza \--p-metric braycurtis \--o-distance-matrix braycurtis_distance_matrix.qza# 可视化和统计分析
qiime diversity alpha-group-significance \--i-alpha-diversity shannon_vector.qza \--m-metadata-file your_sample_metadata.txt \--o-visualization shannon_group_significance.qzv
qiime diversity beta-group-significance \--i-distance-matrix braycurtis_distance_matrix.qza \--m-metadata-file your_sample_metadata.txt \--o-visualization braycurtis_group_significance.qzv-
q2-diversity-lib
q2-diversity-lib 简介
q2-diversity-lib是QIIME 2的一个插件,用于计算多样性指数和样本间的差异。它基于多种生物多样性指标来评估微生物群落的多样性,并允许用户进行统计比较和可视化。
功能特性
- 计算多样性指数:支持计算多种多样性指数,如Shannon、Simpson、Chao1等,可以帮助衡量群落内物种的多样性和丰富度。
- Beta多样性计算:计算不同样本之间的差异和相似性,例如Bray-Curtis、Jaccard、Unweighted UniFrac、Weighted UniFrac等距离指标。
- Beta多样性可视化:生成多样性分析的可视化图表,如PCoA(Principal Coordinates Analysis)图表,展示样本之间的差异。
- 组间比较:支持组间多样性比较,通过PERMANOVA(Permutational Multivariate Analysis of Variance)等方法进行样本群落的差异性分析。
qiime diversity-lib --help
Usage: qiime diversity-lib [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin computes individual metrics for communityalpha and beta diversity.Plugin website: https://github.com/qiime2/q2-diversity-libGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:alpha-passthrough Alpha Passthrough (non-phylogenetic)beta-passthrough Beta Passthrough (non-phylogenetic)beta-phylogenetic-meta-passthroughBeta Phylogenetic Meta Passthroughbeta-phylogenetic-passthrough Beta Phylogenetic Passthroughbray-curtis Bray-Curtis Dissimilarityfaith-pd Faith's Phylogenetic Diversityjaccard Jaccard Distanceobserved-features Observed Featurespielou-evenness Pielou's Evennessshannon-entropy Shannon's Entropyunweighted-unifrac Unweighted Unifracweighted-unifrac Weighted Unifrac
计算使用
#计算Alpha多样性(多样性指数)
qiime diversity alpha \--i-table table.qza \--p-metric shannon \--o-alpha-diversity shannon_alpha.qza#计算Beta多样性距离
qiime diversity beta \--i-table table.qza \--p-metric braycurtis \--o-distance-matrix braycurtis_distance.qza#可视化Beta多样性分析结果(例如PCoA)
qiime diversity pcoa \--i-distance-matrix braycurtis_distance.qza \--o-pcoa braycurtis_pcoa.qza
qiime emperor plot \--i-pcoa braycurtis_pcoa.qza \--m-metadata-file sample-metadata.tsv \--o-visualization braycurtis_emperor.qzv#进行组间多样性比较(以PERMANOVA为例)
qiime diversity beta-group-significance \--i-distance-matrix braycurtis_distance.qza \--m-metadata-file sample-metadata.tsv \--o-visualization braycurtis_permanova.qzv \--p-method permanova
-
q2-emperor
qiime emperor --help
Usage: qiime emperor [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin wraps Emperor and supports interactivevisualization of ordination plots.Plugin website: http://emperor.microbio.meGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:biplot Visualize and Interact with Principal Coordinates AnalysisBiplotplot Visualize and Interact with Principal Coordinates AnalysisPlotsprocrustes-plot Visualize and Interact with a procrustes plot
-
q2-feature-classifier
q2-feature-classifier 是 QIIME 2 中的一个插件,用于对16S rRNA或ITS等序列数据进行分类和注释。它主要用于将序列分类为特定的分类单元,比如对OTUs(操作分类单元)或者物种进行分类。
qiime feature-classifier --help
Usage: qiime feature-classifier [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports taxonomic classification offeatures using a variety of methods, including Naive Bayes, vsearch, andBLAST+.Plugin website: https://github.com/qiime2/q2-feature-classifierGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:blast BLAST+ local alignment search.classify-consensus-blast BLAST+ consensus taxonomy classifierclassify-consensus-vsearch VSEARCH-based consensus taxonomy classifierclassify-hybrid-vsearch-sklearnALPHA Hybrid classifier: VSEARCH exact match+ sklearn classifierclassify-sklearn Pre-fitted sklearn-based taxonomy classifierextract-reads Extract reads from reference sequences.find-consensus-annotation Find consensus among multiple annotations.fit-classifier-naive-bayes Train the naive_bayes classifierfit-classifier-sklearn Train an almost arbitrary scikit-learnclassifiermakeblastdb Make BLAST database.vsearch-global VSEARCH global alignment search
#训练一个 SILVA 数据库上的分类器可以使用如下命令
qiime feature-classifier fit-classifier-naive-bayes \--i-reference-reads silva-132-99-515-806-nb-classifier.qza \--i-reference-taxonomy silva-132-99-515-806-nb-classifier.qza \--o-classifier classifier.qza# 使用训练好的分类器对样本数据进行分类
qiime feature-classifier classify-sklearn \--i-classifier classifier.qza \--i-reads paired-end-demux.qza \--o-classification taxonomy.qza
-
q2-feature-table
q2-feature-table 插件是 QIIME 2 中的一个重要插件,用于处理和操作特征表(feature table)数据,它包括了许多功能,用于对微生物组数据进行分析和可视化。
以下是 q2-feature-table 插件的一些主要功能和使用方法:
-
导入特征表数据: 可以使用该插件将不同格式的特征表数据导入到 QIIME 2 中,如BIOM格式、文本格式、或其他常见格式的特征表。
-
特征表汇总和统计: 可以对特征表进行汇总和统计描述,比如计算每个样本中的特征数量、每个特征在样本中的出现频率等。
-
特征表的过滤和修剪: 提供了多种方法对特征表进行过滤和修剪,如去除低频特征、去除低丰度特征、保留指定样本数或特征数等。
-
特征表的转换和变换: 可以对特征表进行转换,如转置、归一化、对数转换等,以适应不同类型的分析需求。
-
特征表的合并和拆分: 可以将多个特征表合并为一个,也可以根据样本元数据信息将特征表拆分为多个子集。
-
特征表的可视化: 支持对特征表进行可视化展示,比如生成特征数量分布图、绘制热图展示特征在样本中的丰度等。
qiime feature-table --help
Usage: qiime feature-table [OPTIONS] COMMAND [ARGS]...Description: This is a QIIME 2 plugin supporting operations on sample byfeature tables, such as filtering, merging, and transforming tables.Plugin website: https://github.com/qiime2/q2-feature-tableGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:core-features Identify core features in tablefilter-features Filter features from tablefilter-features-conditionally Filter features from a table based onabundance and prevalencefilter-samples Filter samples from tablefilter-seqs Filter features from sequencesgroup Group samples or features by a metadatacolumnheatmap Generate a heatmap representation of afeature tablemerge Combine multiple tablesmerge-seqs Combine collections of feature sequencesmerge-taxa Combine collections of feature taxonomiespresence-absence Convert to presence/absencerarefy Rarefy tablerelative-frequency Convert to relative frequenciesrename-ids Renames sample or feature ids in a tablesplit Split one feature table into manysubsample Subsample tablesummarize Summarize tabletabulate-seqs View sequence associated with each featuretranspose Transpose a feature table.
#特征表的摘要和统计信息
qiime feature-table summarize \--i-table feature-table.qza \--o-visualization feature-table-summary.qzv#特征表的过滤和修剪
qiime feature-table filter-features \--i-table feature-table.qza \--p-min-frequency 10 \--o-filtered-table filtered-feature-table.qzaqiime feature-table filter-samples \--i-table feature-table.qza \--p-min-frequency 500 \--o-filtered-table filtered-sample-table.qza# 特征表的合并和操作
qiime feature-table merge \--i-tables table1.qza \--i-tables table2.qza \--o-merged-table merged-table.qza#计算特征表的β-diversity:
qiime diversity beta \--i-table feature-table.qza \--o-distance-matrix beta-diversity.qza \--p-metric braycurtis#可视化特征表
qiime feature-table summarize \--i-table feature-table.qza \--o-visualization feature-table-summary.qzv#可视化β-diversity距离矩阵
qiime diversity beta-phylogenetic \--i-table feature-table.qza \--i-phylogeny rooted-tree.qza \--o-distance-matrix beta-diversity.qza \--p-metric unweighted_unifracqiime diversity pcoa \--i-distance-matrix beta-diversity.qza \--o-pcoa pcoa-results.qzaqiime emperor plot \--i-pcoa pcoa-results.qza \--m-metadata-file sample-metadata.tsv \--o-visualization emperor.qzv
-
q2-fragment-insertion
q2-fragment-insertion插件是用于将未分配的DNA序列(通常是16S rRNA或18S rRNA序列)嵌入(插入)到预先构建的参考进化树中的工具。这个插件可以帮助解决一些问题,比如通过将未知序列嵌入到进化树中,来推断未知序列的系统发育位置。
qiime fragment-insertion --help
Usage: qiime fragment-insertion [OPTIONS] COMMAND [ARGS]...Description: No description available. See plugin website:https://github.com/qiime2/q2-fragment-insertionPlugin website: https://github.com/qiime2/q2-fragment-insertionGetting user support: https://github.com/qiime2/q2-fragment-insertion/issuesOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:classify-otus-experimental Experimental: Obtain taxonomic lineages, byfinding closest OTU in reference phylogeny.filter-features Filter fragments in tree from table.sepp Insert fragment sequences using SEPP intoreference phylogenies.
qiime fragment-insertion sepp \--i-representative-sequences <代表序列文件.qza> \--i-reference-database <参考数据库.qza> \--o-tree <输出进化树.qza> \--o-placements <输出插入位置文件.qza>
--i-representative-sequences是代表性序列文件的位置。--i-reference-database是参考数据库文件的位置。--o-tree指定输出的进化树文件。--o-placements指定输出的插入位置文件。
-
q2-longitudinal
q2-longitudinal 插件是 QIIME 2 中的一个插件,专门用于处理微生物组长期研究的数据。该插件允许用户对时间序列实验数据进行分析,以便检测微生物组随时间变化的情况,比较不同条件下的变化,以及对这些变化的统计显著性进行评估。
以下是 q2-longitudinal 插件的一些主要功能和使用命令:
主要功能:
- 时间序列数据可视化:生成时间序列样本数据的可视化图表,比如长期研究的变化趋势、样本之间的差异等。
- 差异分析:比较不同时间点或不同处理组之间的微生物组成差异。
- Alpha 和 Beta 多样性分析:评估微生物群落在时间序列中的多样性和相似性变化。
- 线性混合效应模型:对微生物组数据进行线性模型分析,以研究时间、处理效应和其交互作用对微生物组成的影响。
qiime longitudinal --help
Usage: qiime longitudinal [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports methods for analysis of timeseries data, involving either paired sample comparisons or longitudinalstudy designs.Plugin website: https://github.com/qiime2/q2-longitudinalGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:anova ANOVA testfeature-volatility Feature volatility analysisfirst-differences Compute first differences or difference frombaseline between sequential statesfirst-distances Compute first distances or distance from baselinebetween sequential stateslinear-mixed-effects Linear mixed effects modelingmaturity-index Microbial maturity index prediction.nmit Nonparametric microbial interdependence testpairwise-differences Paired difference testing and boxplotspairwise-distances Paired pairwise distance testing and boxplotsplot-feature-volatility Plot longitudinal feature volatility andimportancesvolatility Generate interactive volatility plot
#导入数据
qiime longitudinal feature-table-merge \--i-longitudinal-feature-tables feature-table.qza \--o-merged-table merged-table.qza#线性混合效应模型
qiime longitudinal linear-mixed-effects \--m-metadata-file sample-metadata.tsv \--m-metadata-file timepoints.tsv \--p-metric Shannon_index \--p-group-columns treatment \--p-state-column time \--p-individual-id-column subject \--o-visualization lme-results.qzv#可视化
qiime longitudinal volatility \--i-table merged-table.qza \--m-metadata-file sample-metadata.tsv \--p-state-column time \--p-individual-id-column subject \--o-visualization volatility.qzv#组间差异
qiime longitudinal pairwise-differences \--m-metadata-file sample-metadata.tsv \--m-metadata-file timepoints.tsv \--p-metric Shannon_index \--p-group-column treatment \--p-state-column time \--p-individual-id-column subject \--o-visualization pairwise-differences.qzv#α多样性图表
qiime longitudinal maturity-index \--i-alpha-diversity alpha-diversity.qza \--m-metadata-file sample-metadata.tsv \--p-state-column time \--p-individual-id-column subject \--o-visualization maturity-index.qzv
-
q2-metadata
q2-metadata 插件用于处理和操作元数据,元数据是描述样本信息的数据,比如样本来源、处理方法、实验条件等。以下是 q2-metadata 插件的简要介绍和一些常见的使用命令:
q2-metadata 插件的功能:
- 元数据导入: 将元数据文件导入到 QIIME 2 格式中。
- 元数据可视化: 可视化元数据内容以便更好地理解样本信息。
- 元数据的处理和编辑: 对元数据进行筛选、编辑和转换。
- 元数据统计和摘要: 统计和生成关于元数据的摘要信息。
qiime metadata --help
Usage: qiime metadata [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin provides functionality for working with andvisualizing Metadata.Plugin website: https://github.com/qiime2/q2-metadataGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:distance-matrix Create a distance matrix from a numeric Metadata columnmerge Merge metadatashuffle-groups Shuffle values in a categorical sample metadata column.tabulate Interactively explore Metadata in an HTML table
# 1. 元数据导入:
qiime metadata tabulate --m-input-file sample-metadata.tsv --o-visualization sample-metadata.qzv
# 2. 元数据可视化:
qiime metadata tabulate --m-input-file sample-metadata.qza --o-visualization sample-metadata.qzv
# 3. 元数据编辑与处理:
# 删除列:
qiime metadata tabulate --m-input-file sample-metadata.qza --o-visualization sample-metadata.qzv
# 筛选行(过滤):
qiime metadata tabulate --m-input-file sample-metadata.qza --o-visualization sample-metadata.qzv
# 4. 元数据统计和摘要:
qiime metadata tabulate --m-input-file sample-metadata.qza --o-visualization sample-me-
q2-phylogeny
q2-phylogeny插件则提供了处理生物多样性分析中的系统发育信息的功能。该插件主要用于构建系统发育树和处理系统发育树相关的操作,例如序列的进化树推断、进化树的根节点分配等。
qiime phylogeny --help
Usage: qiime phylogeny [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports generating and manipulatingphylogenetic trees.Plugin website: https://github.com/qiime2/q2-phylogenyGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:align-to-tree-mafft-fasttree Build a phylogenetic tree using fasttree andmafft alignmentalign-to-tree-mafft-iqtree Build a phylogenetic tree using iqtree andmafft alignment.align-to-tree-mafft-raxml Build a phylogenetic tree using raxml andmafft alignment.fasttree Construct a phylogenetic tree with FastTree.filter-table Remove features from table if they're notpresent in tree.filter-tree Remove features from tree based on metadataiqtree Construct a phylogenetic tree with IQ-TREE.iqtree-ultrafast-bootstrap Construct a phylogenetic tree with IQ-TREEwith bootstrap supports.midpoint-root Midpoint root an unrooted phylogenetic tree.raxml Construct a phylogenetic tree with RAxML.raxml-rapid-bootstrap Construct a phylogenetic tree with bootstrapsupports using RAxML.robinson-foulds Calculate Robinson-Foulds distance betweenphylogenetic trees.
## 构建系统发育树:
#使用多序列比对后的结果构建系统发育树。可以使用q2-phylogeny中的FastTree或RAxML进行树的构建。以下是使用FastTree的示例命令:
qiime phylogeny fasttree \--i-alignment aligned-sequences.qza \--o-tree tree.qza# 或者使用RAxML进行系统发育树的构建:
qiime phylogeny raxml \--i-alignment aligned-sequences.qza \--p-substitution-model GTRGAMMA \--o-tree tree.qza \--verbose## 可选操作 - 根节点分配:
# 有时,你可能需要为系统发育树分配根节点。可以使用q2-phylogeny中的根节点分配插件进行此操作。以下是一个示例命令:
qiime phylogeny midpoint-root \--i-tree tree.qza \--o-rooted-tree rooted-tree.qza-
q2-quality-control
q2-quality-control 插件旨在进行序列数据的质量控制和过滤,它可以执行以下任务:
- 对序列数据进行质量评估
- 去除低质量序列
- 截取或修剪序列的部分
- 去除嵌合序列(chimeras)
- 过滤低频序列
-
q2-quality-filter
q2-quality-filter插件用于对DNA测序数据进行质量控制和过滤,以去除低质量的序列。这有助于提高后续分析的准确性和可靠性。
qiime quality-control --help
Usage: qiime quality-control [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports methods for assessing andcontrolling the quality of feature and sequence data.Plugin website: https://github.com/qiime2/q2-quality-controlGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:bowtie2-build Build bowtie2 index from reference sequences.decontam-identify Identify contaminantsdecontam-identify-batches Identify contaminants in Batch Modedecontam-remove Removes contaminantdecontam-score-viz Generate a histogram representation of the scoresevaluate-composition Evaluate expected vs. observed taxonomiccomposition of samplesevaluate-seqs Compare query (observed) vs. reference (expected)sequences.evaluate-taxonomy Evaluate expected vs. observed taxonomicassignmentsexclude-seqs Exclude sequences by alignmentfilter-reads Filter demultiplexed sequences by alignment toreference database.
qiime quality-filter q-score \--i-demux paired-end-demux.qza \--p-min-quality 20 \--o-filtered-sequences demux-filtered.qza \--o-filter-stats demux-filter-stats.qza
-
q2-sample-classifier
而q2-sample-classifier是Qiime 2中的一个插件,用于样本分类和预测。它可以帮助用户利用机器学习算法对样本进行分类,比较不同条件下的微生物组成差异,例如,预测分类数据(如临床数据)和微生物组成之间的关系。
qiime sample-classifier --help
Usage: qiime sample-classifier [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin supports methods for supervisedclassification and regression of sample metadata, and other supervisedmachine learning methods.Plugin website: https://github.com/qiime2/q2-sample-classifierGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:classify-samples Train and test a cross-validated supervisedlearning classifier.classify-samples-from-dist Run k-nearest-neighbors on a labeled distancematrix.classify-samples-ncv Nested cross-validated supervised learningclassifier.confusion-matrix Make a confusion matrix from sample classifierpredictions.fit-classifier Fit a supervised learning classifier.fit-regressor Fit a supervised learning regressor.heatmap Generate heatmap of important features.metatable Convert (and merge) positive numeric metadata(in)to feature table.predict-classification Use trained classifier to predict target valuesfor new samples.predict-regression Use trained regressor to predict target valuesfor new samples.regress-samples Train and test a cross-validated supervisedlearning regressor.regress-samples-ncv Nested cross-validated supervised learningregressor.scatterplot Make 2D scatterplot and linear regression ofregressor predictions.split-table Split a feature table into training and testingsets.summarize Summarize parameter and feature extractioninformation for a trained estimator.
qiime sample-classifier classify-samples \--i-table feature-table.qza \--m-metadata-file sample-metadata.qza \--m-metadata-column TARGET_COLUMN \--p-test-size 0.2 \--p-random-state 42 \--p-n-estimators 100 \--p-n-jobs 1 \--o-visualization classification-results.qzv
--i-table指定特征表格的位置--m-metadata-file指定样本元数据的位置--m-metadata-column指定用于分类的目标列--p-test-size设置测试集的比例--p-random-state设置随机种子以确保结果可重复--p-n-estimators设置分类器使用的估计器数量--p-n-jobs设置用于计算的作业数--o-visualization指定输出结果的位置
-
q2-taxa
q2-taxa 插件则用于分析和可视化分类学信息,特别是对于已经进行了序列分类(比如16S rRNA)的数据。q2-taxa 允许用户对物种注释信息进行处理、汇总和可视化。
qiime taxa --help
Usage: qiime taxa [OPTIONS] COMMAND [ARGS]...Description: This QIIME 2 plugin provides functionality for working with andvisualizing taxonomic annotations of features.Plugin website: https://github.com/qiime2/q2-taxaGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:barplot Visualize taxonomy with an interactive bar plotcollapse Collapse features by their taxonomy at the specified levelfilter-seqs Taxonomy-based feature sequence filter.filter-table Taxonomy-based feature table filter.
# 1. 导入数据
# 使用命令 qiime taxa 可以导入并生成用于分类学分析的数据格式。例如:
qiime taxa import \--input-path taxonomy.tsv \--output-path taxonomy.qza \--type 'FeatureData[Taxonomy]'# 2. 查看物种丰度
# 命令 qiime taxa barplot 可以用于生成物种丰度的柱状图,用于可视化各个样本中不同分类水平的相对丰度。
qiime taxa barplot \--i-table table.qza \--i-taxonomy taxonomy.qza \--m-metadata-file sample-metadata.tsv \--o-visualization taxa-bar-plot.qzv# 3. 物种注释
# 可通过 qiime taxa classify-sklearn 命令使用机器学习分类器进行序列的物种注释。以下是一个例子:
qiime taxa classify-sklearn \--i-reads rep-seqs.qza \--i-classifier classifier.qza \--o-classification taxonomy.qza# 4. 特定分类水平的筛选和可视化
# 可以使用 qiime taxa filter-table 和 qiime taxa filter-seqs 命令在特定分类水平对数据进行过滤。然后,可以使用其他命令可视化已过滤的数据。# 5. 物种多样性分析
使用 qiime taxa collapse 和 qiime taxa rarefy 等命令可以对物种注释数据进行聚合和稀释,以进行多样性分析。-
q2-types
q2-types插件旨在处理和管理不同类型的数据,包括:
- DNA 序列
- RNA 序列
- 蛋白质序列
- 样本和元数据信息
-
q2-vsearch
q2-vsearch 插件则是 QIIME 2 中用于序列相似性搜索和聚类的插件之一。它基于 VSEARCH,提供了执行序列相似性搜索、聚类和序列分析的功能。这个基本上与直接使用vesearch直接使用差不多了,只不过这里集成后可以直接与qiime的其他模块相结合使用。
qiime vsearch --help
Usage: qiime vsearch [OPTIONS] COMMAND [ARGS]...Description: This plugin wraps the vsearch application, and provides methodsfor clustering and dereplicating features and sequences.Plugin website: https://github.com/qiime2/q2-vsearchGetting user support: Please post to the QIIME 2 forum for help with thisplugin: https://forum.qiime2.orgOptions:--version Show the version and exit.--example-data PATH Write example data and exit.--citations Show citations and exit.--help Show this message and exit.Commands:cluster-features-closed-referenceClosed-reference clustering of features.cluster-features-de-novo De novo clustering of features.cluster-features-open-referenceOpen-reference clustering of features.dereplicate-sequences Dereplicate sequences.fastq-stats Fastq stats with vsearch.merge-pairs Merge paired-end reads.uchime-denovo De novo chimera filtering with vsearch.uchime-ref Reference-based chimera filtering with
# 序列相似性搜索
# 使用 q2-vsearch 插件进行序列相似性搜索的命令:
qiime vsearch search-sequences \--i-query query_sequences.qza \--i-reference reference_sequences.qza \--o-results search_results.qza \--o-perc-identity output_identity.qza \--o-failed-sequences failed_sequences.qza
# 此命令将执行一项序列搜索,将查询序列与参考序列集进行比较,并生成包含搜索结果的文件。# 序列聚类
# 利用 q2-vsearch 进行序列聚类的命令示例:
qiime vsearch cluster-features-de-novo \--i-sequences sequences.qza \--i-table table.qza \--p-perc-identity 0.97 \--o-clustered-table clustered_table.qza \--o-clustered-sequences clustered_sequences.qza \--o-new-reference-sequences new_reference_sequences.qza当然还有分析结果的可视化
在线可视化,拖拽出图:QIIME 2 View
相关文章:
基于conda环境使用mamba/conda安装配置QIIME 2 2023.9 Amplicon扩增子分析环境,q2cli主要功能模块介绍及使用
QIIME 2 2023.9 Amplicon Distribution介绍: 概述 qiime团队专门针对高通量扩增子序列分析退出的conda集成环境,包括了主要和常见的扩增子分析模块,用户可以单独使用各个模块,也可以使用各模块组成不同的分析流程。从2023.09版本…...

腾讯-轻量应用服务器centos7中宝塔安装MySQL8.0出现内存不足
目录 前言 出现的问题: 解决方法: 编译安装: 极速安装 其他 我的其他博客 前言 说实话,本人也就是个穷学生买不起啥大的服务器啥的,整了个2核 2内存的服务器 用宝塔按mysql5.5是没问题的,一切换8.0就提醒内存不足…...
调用Win10隐藏的语音包
起因 在做一个文本转语音的Demo的时候,遇到了语音包无法正确被Unity识别的问题。明明电脑上安装了语音包但是代码就是识别不出来 原因 具体也不是非常清楚,但是如果语言包是在的话,大概率是Win10系统隐藏了。 确定语言包 首先查看%windi…...

【WPF】应用程序和已知安卓设备的局域网IP之间进行通信
要在WPF应用程序和已知安卓设备的局域网IP之间进行通信,可以使用Socket通信。以下是一个基本的示例: 在WPF应用程序中创建一个Socket对象并连接到安卓设备的IP地址和端口号: using System.Net.Sockets;// 创建一个Socket对象 Socket socket…...
linux Ubuntu下,第一个C++程序访问数据库,遇到的问题,及解决办法
在ubuntu下安装了mysql,mysql以后,编写了第一个访问数据库的程序: #include <iostream> #include <string> #include <cstdlib> //for system #include <mysql.h>using namespace std;int main() {mysqlpp::Connect…...

【Flink on k8s】- 7 - 在本地运行第一个 flink wordcount job
目录 1、环境准备 2、代码开发 3、启动运行 4、在控制台找到 web ui,查看监控...

velocity-engine-core是什么?Velocity模板引擎的使用
velocity-engine-core是什么?Velocity模板引擎的使用 1. 常见的模板引擎2. Velocity 的语法3.Velocity的使用 相信在日常开发中或多或少都听过或者使用过模板引擎,比如熟知的freemarker, thymeleaf等。而模板引擎就是为了实现View和Data分离而产生的。 而…...

【华为od】存在一个m*n的二维数组,其成员取值范围为0,1。其中值为1的元素具备扩散性,每经过1S,将上下左右值为0的元素同化为1。
存在一个m*n的二维数组,其成员取值范围为0,1。其中值为1的元素具备扩散性,每经过1S,将上下左右值为0的元素同化为1。将数组所有成员初始化为0,将矩阵的[i, j]和[m,n]位置上元素修改成1后,在经过多长时间所有元素变为1。 输入描述 输入的前两个数字是矩阵大小。后面是数字…...
docker学习(七、搭建mysql8.2主从)
一、主库搭建 1.构建主库镜像 # 运行mysql镜像,配置端口3307为主库 docker run -p 3307:3306 --name mysql-master --privilegedtrue -v /mydata/mysql-master/log:/var/log/mysql -v /mydata/mysql-master/data:/var/lib/mysql -v /mydata/mysql-master/conf:/etc…...

消费升级:无人零售的崛起与优势
消费升级:无人零售的崛起与优势 随着人们生活水平的提高,消费内容正在从生存型消费转向以精神体验和享乐为主的发展型消费。社会居民的消费结构不断变迁,明显呈现消费升级趋势。个性化和多元化消费势头正在崛起,特别是无人零售的自…...

【开题报告】基于SpringBoot的煤炭企业安全宣传学习平台的设计与实现
1.选题背景 煤炭企业作为我国能源行业的重要组成部分,承担着国民经济的支撑和推动作用。然而,煤炭生产过程中存在较高的安全风险,煤矿事故频发,给人员生命财产安全带来严重威胁,也给社会稳定和经济发展带来不利影响。…...

机器连接和工业边缘计算
软件应用和IT创新是制造业投资的主要驱动力。解决方案架构应围绕特定标准进行整合,并采用架构蓝图和最佳实践来满足最终用户的需求。此外,边缘计算(Edge Computing)也将在制造业中加速部署。 边缘计算是制造业的下一个变革驱动力。…...
java系列-LinkedHashMap
1.插入新节点时,会将该节点加到链表尾部 public class LinkedHashMap<K,V> extends HashMap<K,V> implements Map<K,V>{/*** The head (eldest) of the doubly linked list.*/transient LinkedHashMapEntry<K,V> head;/*** The tail (young…...

【linux】查看CPU和内存信息
之前咱们一起学习了查看内存的和CPU的命令。 mpstat : 【linux】 mpstat 使用 uptime:【Linux】 uptime命令使用 CPU的使用率:【linux】查看CPU的使用率 nmon :【linux】nmon 工具使用 htop :【linux】htop 命令…...
【产品经理】产品专业化提升路径
产品专业化就是上山寻路,梳理一套作为产品经理的工作方法。本文作者从设计方法、三基座、专业强化、优秀产品拆解、零代码这五个方面,对产品经理的产品专业化进行了总结归纳,一起来看一下吧。 产品专业化就是上山寻路,梳理一套作为…...
卸载与安装指定版本的 openssl)
Ubuntu(WSL)卸载与安装指定版本的 openssl
卸载 openssl 1)查找并删除 openssl 关联的目录与文件 whereis opensslwhich opensslrm -rf /a/b/c/ // 使用 rm 命令删除所有 openssl 相关目录 2)删除软件安装包 apt-get purge openssl 3)删除配置文件 rm -rf /etc/ssl 安装 ope…...

leetcode1115. 交替打印 FooBar
题目 1115. 交替打印 FooBar 给你一个类: class FooBar {public void foo() {for (int i 0; i < n; i) {print("foo");}}public void bar() {for (int i 0; i < n; i) {print("bar");}} }两个不同的线程将会共用一个 FooBar 实例&am…...

qt有哪些常用控件
Qt 是一个跨平台的应用程序开发框架,提供了许多不同类型的控件来构建用户界面。以下是一些常见的 Qt 控件: 按钮(Button):用于执行操作或触发事件。文本框(TextBox):用于输入和显示文…...
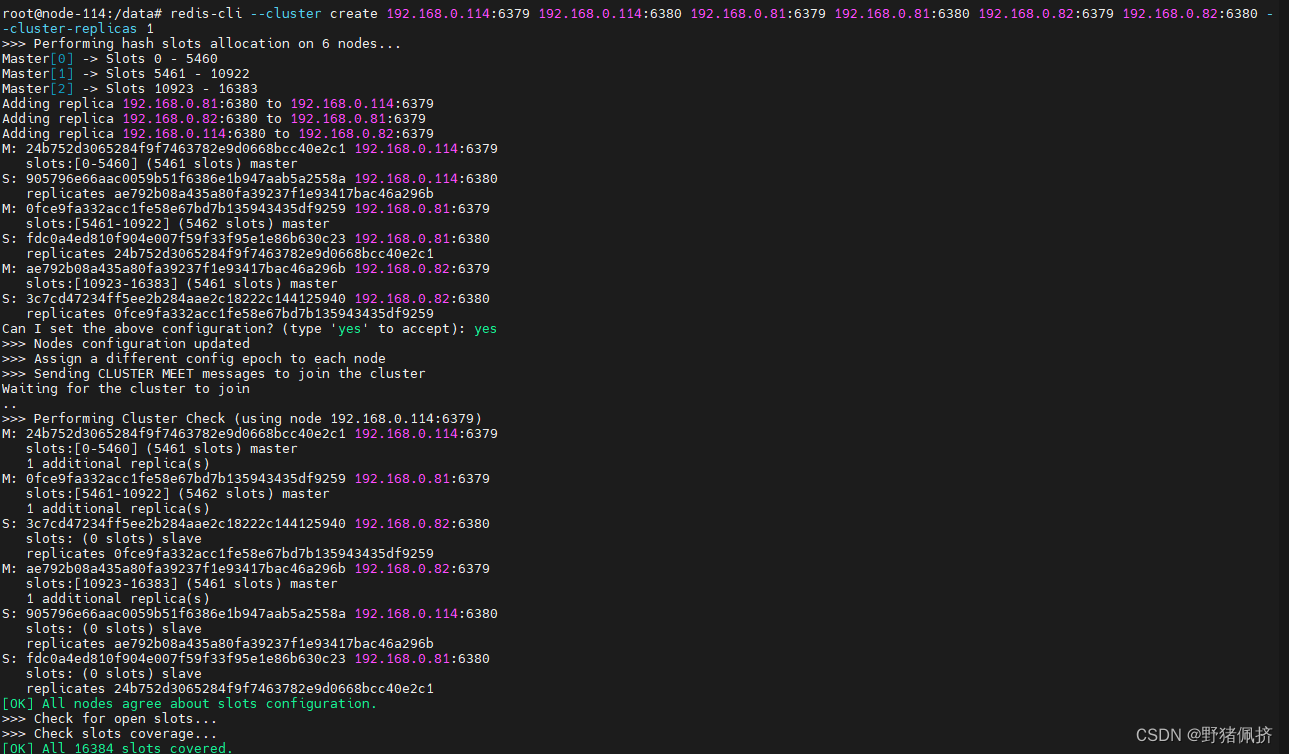
docker 手工redis7.x cluster
IP端口192.168.0.816379/6380192.168.0.826379/6380192.168.0.1146379/6380 mdkir /data/{6379,6380}cat <<END> /data/6379.conf # 端口号 port 6379# 设置客户端连接后进行任何其他指定前需要使用的密码 #requirepass 123456 ## 当master服务设置了密码保护时(用re…...

【华为OD题库-082】TLV解析II-Java
题目 两端通过TLVQ格式的报文来通信,现在收到对端的一个TLV格式的消息包,要求生成匹配后的(tag,length,valueOffset)列表。具体要求如下: (1)消息包中多组tag、length、value紧密排列,其中tag,length各占1字节(uint8),value所占字节数等于len…...

【C语言】C语言之数字金字塔进阶实战:从基础到复杂
1. 数字金字塔基础实现 第一次接触数字金字塔时,我被它整齐的排列方式深深吸引。这种通过数字构建的图形不仅美观,更是理解循环结构的绝佳案例。让我们从一个最基础的5层金字塔开始: #include<stdio.h> int main() {int n 5;for(int i…...

如何选择最适合的自动化技术栈:FlaUI与UIA2/UIA3深度解析
如何选择最适合的自动化技术栈:FlaUI与UIA2/UIA3深度解析 【免费下载链接】FlaUI UI automation library for .Net 项目地址: https://gitcode.com/gh_mirrors/fl/FlaUI FlaUI作为一款强大的.NET UI自动化库,提供了对UIA2和UIA3两种技术标准的支持…...

如何突破游戏外设限制?ViGEmBus虚拟手柄驱动技术全攻略
如何突破游戏外设限制?ViGEmBus虚拟手柄驱动技术全攻略 【免费下载链接】ViGEmBus Windows kernel-mode driver emulating well-known USB game controllers. 项目地址: https://gitcode.com/gh_mirrors/vi/ViGEmBus 在游戏世界中,硬件兼容性问题…...

Qwen2.5-VL底座+lychee-rerank-mm效果惊艳:批量图片智能打分可视化展示
Qwen2.5-VL底座lychee-rerank-mm效果惊艳:批量图片智能打分可视化展示 1. 项目简介 这是一个专门为RTX 4090显卡(24G显存)打造的智能图片排序系统。核心基于阿里通义千问Qwen2.5-VL多模态大模型,结合Lychee-rerank-mm专业重排序…...

【书生·浦语】internlm2-chat-1.8b部署避坑指南:模型拉取失败/响应延迟/中文乱码全解
【书生浦语】internlm2-chat-1.8b部署避坑指南:模型拉取失败/响应延迟/中文乱码全解 1. 环境准备与快速开始 在开始使用internlm2-chat-1.8b之前,我们先来了解一下这个模型的基本情况。这是一个拥有18亿参数的中文对话模型,专门针对聊天场景…...

MiniCPM-V-2_6高级教程:C语言文件操作实现批量图片处理流水线
MiniCPM-V-2_6高级教程:C语言文件操作实现批量图片处理流水线 你是不是经常遇到这样的场景:电脑里存了几百上千张图片,需要逐一分析里面的内容,比如识别商品、统计信息或者分类归档?一张张手动处理,不仅效…...
Qwen-Image-2512-Pixel-Art-LoRA 生成像素画音效可视化波形图
Qwen-Image-2512-Pixel-Art-LoRA:当像素画“听见”声音 你有没有想过,声音也能被“画”出来?不是那种抽象的频谱图,而是充满想象力的像素画。最近,我尝试用Qwen-Image-2512模型,结合一个像素艺术风格的LoR…...

SEO宣传推广公司如何做好移动端优化
SEO宣传推广公司如何做好移动端优化 在当前数字化营销的浪潮中,移动端优化已经成为了每一个SEO宣传推广公司必须要掌握的技能之一。随着越来越多的用户通过手机浏览网站和进行在线购物,如何在移动端上获得更高的流量和转化率成为了企业竞争的关键。SEO宣…...

忍者像素绘卷保姆级教程:微信小程序云开发+Serverless函数调用忍者API
忍者像素绘卷保姆级教程:微信小程序云开发Serverless函数调用忍者API 1. 项目介绍与准备工作 忍者像素绘卷是一款基于Z-Image-Turbo深度优化的图像生成工具,它将传统忍者文化与16-Bit复古游戏美学完美融合。本教程将带你从零开始,使用微信小…...

别只盯着代码了!用Multisim仿真带你理解74LS90和555的‘数字心脏’
用Multisim仿真揭秘数字电路:从555脉冲到74LS90计数的实战之旅 当你第一次看到数字电路时,是否觉得那些密密麻麻的芯片引脚和抽象的逻辑符号令人望而生畏?作为一名软件开发者,我曾经也有同样的困惑——直到发现Multisim这个神奇的…...