inferCNV:scRNA-seq数据推断染色体拷贝数变化
inferCNV分析简介
inferCNV用于探索肿瘤单细胞RNA-Seq 数据,以确定体细胞大规模染色体拷贝数改变的证据,例如整个染色体或大片段染色体的增益或缺失。这是通过与一组参考“正常”细胞(这里的正常细胞可自行定义)进行比较,探索肿瘤基因组各部位基因的表达强度来完成的。热图展示每个染色体的相对表达强度,并且与“正常”细胞相比,肿瘤基因组的哪些区域过表达或降低(异常)。
同时我们需要了解两个概念:拷贝数变异(Copy number variation, CNV) 和 拷贝数改变(copy number alterations, CNA) 。
拷贝数改变(copy number alterations, CNA) :指的是基因组中的 DNA 片段在不同个体间的拷贝数存在变异。这些变异可以是片段的缺失(deletion)或扩增(duplication),并且这些片段通常较大,范围从 1 kb 到数 Mb 不等。
拷贝数改变(copy number alterations, CNA) :是指基因组中某些 DNA 片段的拷贝数发生变化,包括拷贝数的增加(扩增,amplification)和减少(缺失,deletion)。这些改变可以影响单个基因、多个基因甚至整个染色体区域, CNA 与CNV概念相似。
inferCNV分析的意义
我们对单细胞分析的时候会根据关键基因/标记进行细胞分群,基本上可以把大部分的细胞亚群给区分开来,但如果某些基因/标记广泛地存在于多种细胞亚群时,此时应进一步寻找其他的关键基因/标记进行区分,这种分析策略是“常规流程”。对于肿瘤细胞和正常细胞而言,我们常会遇到某些关键基因/标记在两者中均表达的情况(转录组水平),此时通过“常规流程”仍难以区分细胞的良恶性,因此换个角度来辅助区分良恶性细胞就显得尤为重要。
肿瘤细胞通常具有更多的CNV,而正常细胞则较为稳定,因此如果有一种工具能够可视化拷贝数变异的情况那么是不是就可以辅助鉴别良恶性细胞呢?基于这种情况inferCNV就应运而生了。
inferCNV分析结果详解
1、官方示例展示

图中的红色部分是References(Cells),也就是我们自行定义的对照(正常)细胞。这些对照细胞可以是相对于肿瘤细胞的正常细胞(癌与非癌),也可选用免疫细胞进行对照。
蓝色部分是Observations(Cells),也就是我们想要重点分析的细胞。
灰色部分是细胞的图注,上边的色块代表了对照细胞的情况,该图研究者纳入了Microglia/Macrophage和Oligodendrocytes(non-malignant)作为对照细胞,恶性细胞作为观察细胞。

红色方框部分代表的是分层聚类树,其中第一列色柱代表的是所有观察组细胞的分层聚类,第二列色柱则是与所提供输入的观察组细胞分类相对应。

第一列色柱根据分析时group_by_cluster参数设置的不同(True/False)会对结果产生差异:
如果group_by_cluster = FALSE ,意味着不按照研究者命名的cluster去分,换句话说就是第一列色柱是按照聚类树所切割成k_obs_groups分组的情况来表示树状图的细分。
如果group_by_cluster = TRUE ,左边的树状图是所有观察细胞的“线性连接”(树状图的根与每种类型的树状图的根相连,这导致它们都处于同一水平)。第一列色柱列是单一颜色,因为注释聚类时不使用k_obs_groups分组。
但实际函数中参数名称改成了cluster_by_groups,并且True/False的最终结果也对调过来了,这个可能跟R包更新了有关系,因此我个人意见是,只要看到第一条色柱是同一的颜色时,我们应当理解为不采用k_obs_groups分组,而当显示不同颜色时是采用了k_obs_groups分组。两种参数设置均可。
蓝色方框部分代表的是染色体的分组分别是从chr1-chr22,性染色体和线粒体染色体已经被过滤了。
红色和蓝色箭头分别代表染色体区域扩增和染色体区域缺失(两者均为异常)。

按照箭头看向对照和观察组细胞,在红色箭头处代表了malignant_MGH36细胞群,这群细胞相比于观察组(蓝色箭头) 在chr1, 4, 19中存在更显著的染色体缺失,在chr11, 21中存在更显著的染色体扩增。同样的我们可以看其他的三群恶性细胞,也均存在不同区域的染色体拷贝数异常。
2、其他数据展示—正常/肿瘤样本
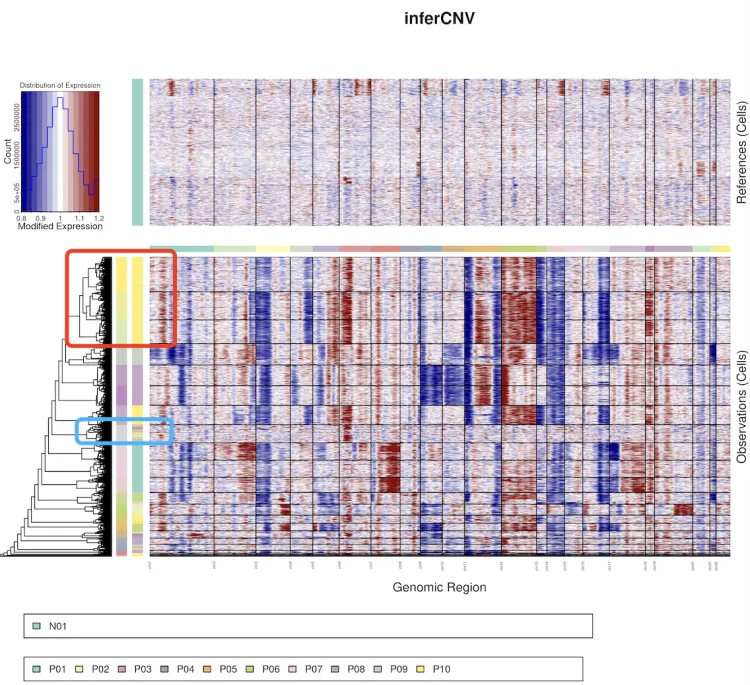
该结果显示1个正常样本作为对照组,10个肿瘤样本作为观察组。整体情况观察下来可以发现肿瘤组样本相比于对照组来说还是存在了很明显的染色体拷贝数异常。
红色方框中的第二列色柱是代表了P10肿瘤样本的信息,中间还混了一小部分的P05患者的信息,从聚类树分层后的色柱来看,P10肿瘤样本还可以进一步的细分为三群。
蓝色方框中的第二列色柱带了很多肿瘤样本的信息,但从聚类树分层后的色柱来看,这些肿瘤样本信息应当分成同一群。

同样的数据,修改了cluster_by_groups的值之后聚类树的图形出现了变化。在不按照k_obs_groups分组之后,主要根据每个样本内部的情况进行分组,我们可以明显发现不同样本内部也存在着很大的异质性(红色方框)。
3、其他数据展示—细胞亚群

该结果显示前期分析得到了17个细胞亚群,其中第15个亚群根据关键基因/标记推测认为可能是正常细胞群,因此通过inferCNV进行辅助分析判断第15个亚群是否是正常细胞群,从结果来看确实也符合正常细胞亚群的猜测。
而根据聚类树结果来看,k_obs_groups分组和细胞亚群分组一致。
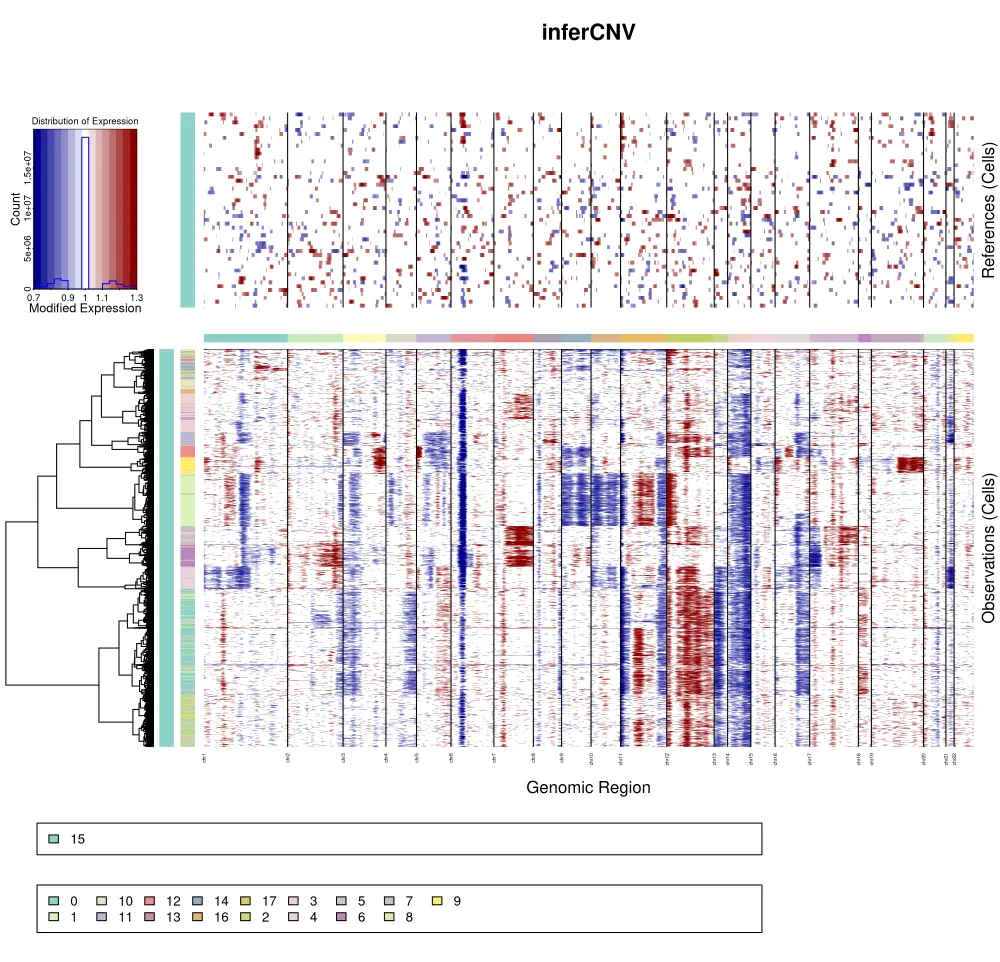
在不按照k_obs_groups分组之后,我们可以明显发现很多细胞亚群之间没有十分明显的界限,也就是说这个结果提示我们在后续聚类的时候可以把很多细胞亚群合并成同一群。
inferCNV分析流程
1.加载对象和R包
rm(list = ls())
library(infercnv)
library(Seurat)
library(gplots)
library(ggplot2)
#这里加载的是seurat对象,替换自己的数据即可
load("sce_CNV_N_T.Rdata") #检查一下自己导入进来的数据
DimPlot(sce,reduction = 'umap',label = TRUE,pt.size = 0.5) +NoLegend()
2.读取数据
#根据需求去检查一下数据集的信息
table(Idents(sce))
table(sce@meta.data$seurat_clusters)
table(sce@meta.data$orig.ident)
#table(sce@meta.data$patient)
#table(sce@meta.data$ID)# 文件制作1:表达量矩阵
# 除了用GetAssayData函数其实也可以直接sce@assays$RNA@count即可
dat <- GetAssayData(sce,layer = 'counts',assay = 'RNA')
dat[1:4,1:4]# 文件制作2:样本的描述
groupinfo <- data.frame(v1 = colnames(dat),v2 = sce@meta.data$patient)# 文件制作3:基因在染色体中的坐标
library(AnnoProbe)
#annoGene 函数返回一个数据框,包含输入基因的详细注释信息。
#注释信息可能包括基因名称、染色体位置、基因描述等。
geneInfor <- annoGene(rownames(dat),"SYMBOL","human") #物种需要小心哦
# 使用逻辑条件来移除包含 chrM, chrX, 和 chrY 的行
geneInfor <- geneInfor[!geneInfor$chr %in% c("chrM", "chrX", "chrY"), ]
#提取chr后后面的数字并转化为num,从而按这个num排序
#sub函数用于把chr替换为空
geneInfor$chr_num <- as.numeric(sub("chr", "", geneInfor$chr))
colnames(geneInfor)#with函数的作用是简化写法,可问gpt
geneInfor <- geneInfor[with(geneInfor,order(chr_num,start)),c(1,4:6)]
geneInfor <- geneInfor[!duplicated(geneInfor[,1]),]length(unique(geneInfor[,1]))
head(geneInfor)
3.排序一下
#保留行名在geneInfor第一列中存在的行。
dat <- dat[rownames(dat) %in% geneInfor[,1],]
#match(x, table):match函数返回x中每个元素在table中的位置索引。
#获得位置后对dat进行重新排序使其跟geneInfor中的顺序一致
dat <- dat[match(geneInfor[,1],rownames(dat)),]
#检查信息
dim(dat)
head(groupinfo)
dat[1:4,1:4]
table(groupinfo$v2)
dim(groupinfo)
4.保存/输出文件
#统一把”-“改成”_“,因为后续运行inferCNV的时候需要读取保存文档,若不修改则会出现错误。
#expFile:是一个变量,存储写入的文件的文件名或路径。在这里文件名是expFile.txt。
expFile <- 'expFile.txt' #定义输出文件名
colnames(dat) <- gsub("-", "_", colnames(dat))
write.table(dat, file = expFile, sep = '\t', quote = F)#groupFiles:是一个变量,存储写入的文件的文件名或路径。在这里文件名是groupFiles.txt。
groupFiles <- 'groupFiles.txt'
groupinfo$v1 <- gsub("-", "_", groupinfo$v1)
write.table(groupinfo,file = groupFiles, sep = '\t',quote = F, col.names = F, row.names = F)#geneFile:是一个变量,存储写入的文件的文件名或路径。在这里文件名是geneFile.txt。
head(geneInfor)
geneFile <- 'geneFile.txt'
write.table(geneInfor, file = geneFile, sep = '\t',quote = F, col.names = F, row.names = F)
5. inferCNV分析
#创建对象,请注意文件需要一一对应哦!
#ref_group_names 这里的细胞是正常对照,然后跟其他的细胞比较
infercnv_obj <- CreateInfercnvObject(raw_counts_matrix = expFile,annotations_file = groupFiles,delim = "\t",gene_order_file = geneFile,ref_group_names = c("N01")
)infercnv_obj2 <- infercnv::run(infercnv_obj,cutoff = 0.1, #smart-seq选择1,10X选择0.1out_dir = "infercnv_output", # dir is auto# 是否根据细胞注释文件的分组# 对肿瘤细胞进行分组# 影响read.dendrogram, 如果有多个细胞类型,且设置为TRUE,# 后续的read.dendrogram无法执行cluster_by_groups = F, #是否根据患者类型(由细胞注释文件中定义)hclust_method = "ward.D2",# ward.D2 方法进行层次聚类analysis_mode = "samples", # 默认是samples,推荐是subclustersdenoise = TRUE, # 去噪音HMM = F, ##特别耗时间,是否要去背景噪音plot_steps = F, #不在每个步骤后生成图形。leiden_resolution = "auto", #可以手动调参数num_threads = 4 #4线程工作,加快速度)

接下来使用曾老师的方法去检查拷贝数变异的情况
6、读取数据加载R包
rm(list = ls())
options(stringsAsFactiors = F)
library(phylogram)
library(gridExtra)
library(grid)
require(dendextend)
require(ggthemes)
library(tidyverse)
library(Seurat)
library(infercnv)
library(miscTools)load("sce_CNV_N_T.Rdata")
7、把inferCNV中的run.final.infercnv_obje文件读取进来
infer_CNV_obj<-readRDS('../3.inferCNV_N_T/infercnv_output/run.final.infercnv_obj')
expr <- infer_CNV_obj@expr.data
expr[1:4,1:4]
data_cnv <- as.data.frame(expr)
dim(expr)
colnames(data_cnv)
rownames(data_cnv)#记得要改一下sce中的列名
colnames(sce) <- gsub("-","_",colnames(sce))
meta <- sce@meta.data
8、计算CNV并绘图
#要根据参数修改哦,我这里的对照组只有N01
if(T){tmp1 = expr[,infer_CNV_obj@reference_grouped_cell_indices$N01]tmp = tmp1#tmp2 = expr[,infer_CNV_obj@reference_grouped_cell_indices$`ref-2`]#tmp= cbind(tmp1,tmp2)down=mean(rowMeans(tmp)) - 2 * mean( apply(tmp, 1, sd))up=mean(rowMeans(tmp)) + 2 * mean( apply(tmp, 1, sd))oneCopy=up-downoneCopya1= down- 2*oneCopya2= down- 1*oneCopydown;upa3= up + 1*oneCopya4= up + 2*oneCopy cnv_score_table<-infer_CNV_obj@expr.datacnv_score_table[1:4,1:4]cnv_score_mat <- as.matrix(cnv_score_table)# Scoringcnv_score_table[cnv_score_mat > 0 & cnv_score_mat < a2] <- "A" #complete loss. 2ptscnv_score_table[cnv_score_mat >= a2 & cnv_score_mat < down] <- "B" #loss of one copy. 1ptscnv_score_table[cnv_score_mat >= down & cnv_score_mat < up ] <- "C" #Neutral. 0ptscnv_score_table[cnv_score_mat >= up & cnv_score_mat <= a3] <- "D" #addition of one copy. 1ptscnv_score_table[cnv_score_mat > a3 & cnv_score_mat <= a4 ] <- "E" #addition of two copies. 2ptscnv_score_table[cnv_score_mat > a4] <- "F" #addition of more than two copies. 2pts# Checktable(cnv_score_table[,1])# Replace with score cnv_score_table_pts <- cnv_score_matrm(cnv_score_mat)# cnv_score_table_pts[cnv_score_table == "A"] <- 2cnv_score_table_pts[cnv_score_table == "B"] <- 1cnv_score_table_pts[cnv_score_table == "C"] <- 0cnv_score_table_pts[cnv_score_table == "D"] <- 1cnv_score_table_pts[cnv_score_table == "E"] <- 2cnv_score_table_pts[cnv_score_table == "F"] <- 2cnv_score_table_pts[1:4,1:4]str(as.data.frame(cnv_score_table_pts[1:4,1:4])) cell_scores_CNV <- as.data.frame(colSums(cnv_score_table_pts))colnames(cell_scores_CNV) <- "cnv_score"
}#可视化
head(cell_scores_CNV)
score=cell_scores_CNV
head(score)
meta$totalCNV = score[match(colnames(sce),rownames(score)),1]
p <- ggplot(meta, aes(x= patient, y=totalCNV, fill=patient)) +geom_boxplot();print(p)
ggsave("totalCNV.png",plot = p, width = 8,height = 6,dpi = 300)

补充资料:
官方说明:
https://github.com/broadinstitute/inferCNV/wiki
技能树推文及B站视频:
推文名称:肿瘤单细胞转录组拷贝数分析结果解读和应用
https://www.bilibili.com/video/BV19Q4y1R7cu/?spm_id_from=333.999.0.0&vd_source=3a13860df939bc922ad1fd6099e42c1d
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员(部分代码来源:生信技能树马拉松和数据挖掘课程)。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(希望多多交流)。更多内容可关注公众号:生信方舟
- END -
相关文章:
inferCNV:scRNA-seq数据推断染色体拷贝数变化
inferCNV分析简介 inferCNV用于探索肿瘤单细胞RNA-Seq 数据,以确定体细胞大规模染色体拷贝数改变的证据,例如整个染色体或大片段染色体的增益或缺失。这是通过与一组参考“正常”细胞(这里的正常细胞可自行定义)进行比较…...

银河麒麟操作系统通过首批软件供应链安全能力认证
麒麟软件产品供应链安全能力获双重肯定!5月30日,经北京赛迪认证中心评估,银河麒麟高级服务器操作系统V10和银河麒麟桌面操作系统V10成为首批获得软件供应链安全能力认证产品,并在操作系统类产品中名列前茅。 软件供应链安全能力评…...

【MySQL】数据库介绍|数据库分类|MySQL的基本结构|MySQL初步认识|SQL分类
目录 数据库介绍 什么是数据库 数据库分类 1.关系型数据库(RDBMS): 2.非关系型数据库: MySQL要学啥 MySQL初步认识 SQL分类 💡推荐 前些天发现了一个巨牛的人工智能学习网站,通俗易懂,风…...

2024年6月11日 (周二) 叶子游戏新闻
万能嗅探: 实测 网页打开 某视频号、某音、某红薯、某站,可以做到无水印的视频和封面下载功能哦,具体玩法大家自行发挥吧。 WPS免登录一键修改器: 去除烦人的登录且能正常使用 日本一首部游戏《拼图世界》上架Steam 30年PS名作日本游戏厂商日本一的首部品…...
JavaSE----类和对象(中)
5. 对象的构造及初始化 5.1 如何初始化对象 通过前面知识点的学习知道,在Java方法内部定义一个局部变量时,必须要初始化,否则会编译失败。 public static void main(String[] args) {int a;System.out.println(a); }// Error:(26, 28) jav…...
STC8增强型单片机进阶开发--OLED显示器(SPI)
系列文章目录 提示:这里可以添加系列文章的所有文章的目录,目录需要自己手动添加 例如:第一章 Python 机器学习入门之pandas的使用 提示:写完文章后,目录可以自动生成,如何生成可参考右边的帮助文档 文章目…...

在CSS中,可以使用 float 属性来设置元素浮动
在CSS中,可以使用float属性来设置元素浮动。float属性有三个值:left、right和none。 float: left;:将元素浮动到左侧。float: right;:将元素浮动到右侧。float: none;:取消元素的浮动(默认值)。…...
wordpress主题开发
科普一:wordpress 是一套用 php 这个语言写的CMS后台管理系统,即我们大家的 wordpress 网站后台是一样的,能体现我们网站外观不同的地方就在于wordpress主题(即皮肤),而这个主题的基本构成是 htmlcssjavasc…...
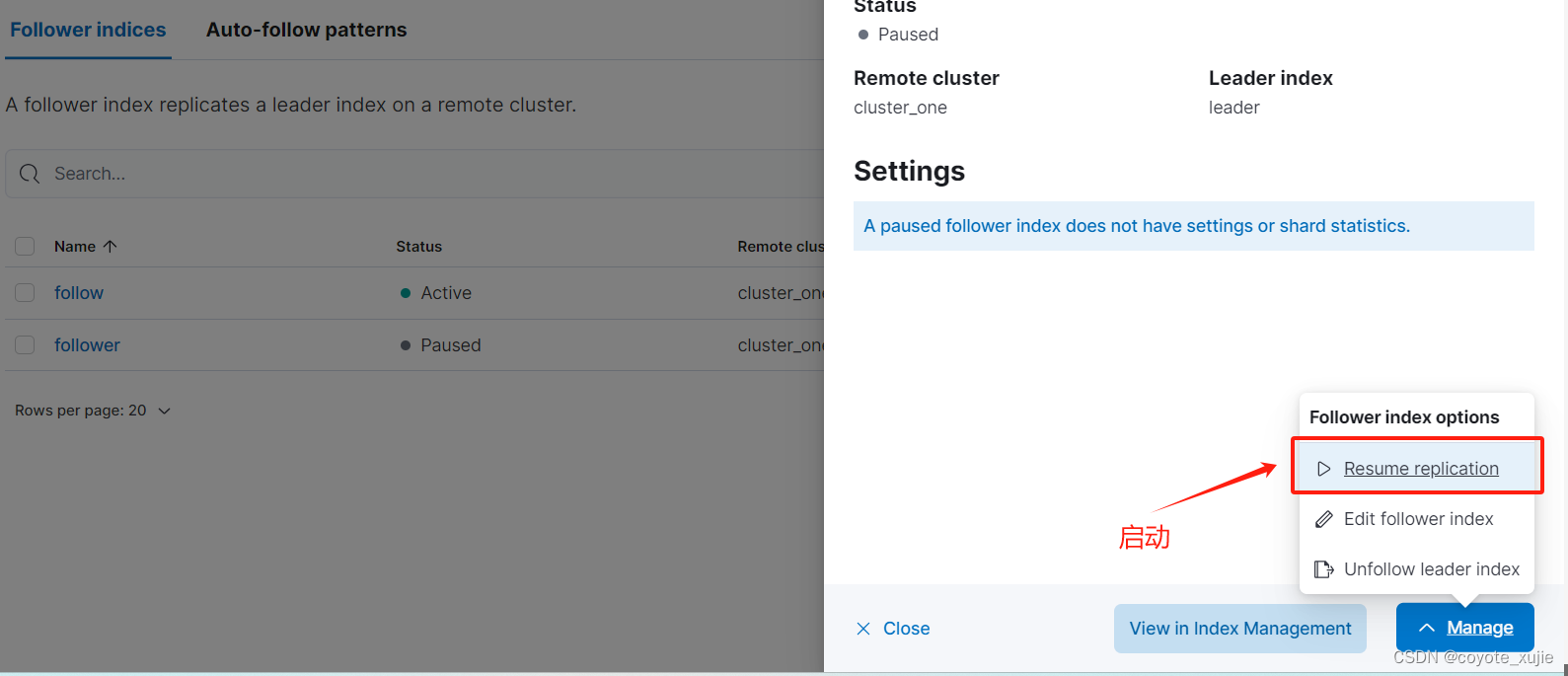
Elasticsearch 认证模拟题 - 17
这两道题目非常具有代表性,分别是跨集群复制和跨集群检索,需要相应的 许可 这里在虚拟机上搭建集群完成这两道题目,这里补充一下 elasticsearch 和 kibana 的配置文件 # elasticsearch.yml cluster.name: cluster2 node.name: cluster2-node…...

Swift 中更现代化的调试日志系统趣谈(一)
概述 昨天凌晨苹果刚刚发布了 WWDC2024 一系列新视频,这标志着苹果开发的一只脚已迈入人工智能(Apple Intelligence)的崭新时代。即便如此,我相信不少秃头码农们还在使用一些“远古简陋”的调试方法来剖析 2142 年的代码。 不过别担心,这一切将在小伙伴们学完本系列博文后…...

深入理解Java中的SPI机制
1. 简介 SPI(Service Provider Interface) 是Java提供的一种为服务框架提供服务实现的机制。它允许框架在运行时动态地发现服务的实现,从而实现模块化设计。在Java中,SPI机制主要用于解耦API和实现,使得应用程序可以在…...

2、python 基础学习总结
文章目录 一、python 标识符和变量命名规则1、python 标识符2 python 变量和变量命名规则 二、数据类型2.1 Numbers(数字类型)2.2 String(字符串类型)2.2.1 单引号、双引号、三引号字符串之间的区别2.2.2 转义字符 在这里插入图片…...

线程的状态!!!
NEW:(初始状态) 线程对象已经创建,但尚未启动。此时,线程还没有开始执行。 RUNNABLE:(运行状态) 线程已经启动并且正在运行,或者准备好运行,但可能由于其他线…...
是什么?如何解决?)
Hsah碰撞(冲突)是什么?如何解决?
Hash冲突:两个不同的对象经过hash计算后得到的hash值相同,导致冲突。 解决方法: 1、开放地址法:在哈希表中寻找其他的空闲位置来存储冲突的元素。 2、拉链法:拉链法的基本思路是在每个哈希槽中存储一个链表。当发生…...

doc 和 docx 文件的区别
人不走空 🌈个人主页:人不走空 💖系列专栏:算法专题 ⏰诗词歌赋:斯是陋室,惟吾德馨 目录 🌈个人主页:人不走空 💖系列专栏:算法专题 ⏰诗词歌…...
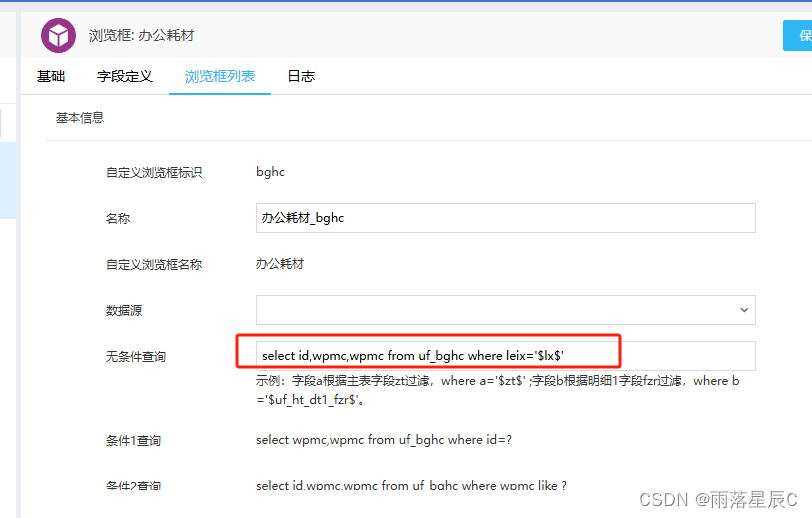
泛微OA E9 浏览框显示的数据根据表单字段过滤
一、实现效果:如图所示,字段“物品名称”浏览框显示的数据根据“类型”字段进行过滤。 二、实现方法: 1、建模引擎-应用建模-浏览框-浏览框列表中单击“办公耗材”-“浏览框列表”-“操作”-“编辑” 2、sql语句中根据OA自带是示例增加where…...
)
AIGC涉及到的算法(一)
目录 1. 生成对抗网络(GAN) 2. 变分自编码器(VAE) 3. 扩散模型(Diffusion Model) 4. Transformer 模型 5. 自然语言处理算法(NLP) 6. 计算机视觉算法(CV) 7. 神经网络算法 8. 决策树算法 9. 遗传算法 10. 聚类算法 1. 生成对抗网络(GAN) 原理与应用:生成对…...

一种基于单片机的智能饮水机设计
随着人们生活水平的提高,对美好生活质量的追求也越来越高。饮 水机是人们日常生活不可或缺的,实现饮水机的智能化控制不但方便, 而且更加安全。本文提出一种基于单片机的智能饮水控制系统,通过传 感器实现对水温的监测,…...
)
竞争性谈判和竞争性磋商的区别(电子化招采系统)
竞争性谈判和竞争性磋商在政府采购和项目采购中都是常用的方式,但它们在多个方面存在显著的区别,郑州信源数智化招采系统可满足各种招标和采购方式,结合多年招采系统研发和实施经验,对竞争性谈判和竞争性磋商的区别总结如下: 1、…...
STM32F413 STM32F423数据手册 中文版 STM32F413 STM32F423勘误手册英文版等文档
链接: https://pan.baidu.com/s/1AeYaoFb5Wurii6OM2ZlY2Q 提取码: a3tj 本文分享关于STM32F413 和STM32F423芯片的相关资料,主要资源如下图所示: 包含的文档有: STM32F40xxx and STM32F41xxx单片机编程手册 中文版 英文版 STM32F413xG 423…...

Windows升级Node版本指南
在 Windows 上升级 Node.js,主要有四种方法,各有侧重。对于大多数开发者,使用版本管理工具 nvm-windows 是最灵活高效的选择。 Windows安装Node.js: 步骤1:访问 Node.js 官方网站 官方网站,下载适用于 Wind…...
Loop Habit Tracker习惯追踪应用技术深度解析与架构实践指南
Loop Habit Tracker习惯追踪应用技术深度解析与架构实践指南 【免费下载链接】uhabits Loop Habit Tracker, a mobile app for creating and maintaining long-term positive habits 项目地址: https://gitcode.com/gh_mirrors/uh/uhabits Loop Habit Tracker是一款基于…...

摄像头驱动调试避坑指南:用示波器快速定位I2C不通、MIPI无信号问题
摄像头驱动调试避坑指南:用示波器快速定位I2C不通、MIPI无信号问题 当摄像头模组在硬件调试阶段出现异常时,软件工程师往往会陷入"配置检查-重新烧录-再检查"的死循环。实际上,80%的摄像头初始化失败问题源于硬件信号层面的异常。本…...

如何用JPlag守护代码原创性:5分钟快速上手指南
如何用JPlag守护代码原创性:5分钟快速上手指南 【免费下载链接】JPlag State-of-the-Art Source Code Plagiarism & Collusion Detection. Check for plagiarism in a set of programs. 项目地址: https://gitcode.com/gh_mirrors/jp/JPlag 你是否曾担心…...

从 ROS 到 Cognitive OS、Agentic OS:机器人操作系统与具身智能新时代
一、先搞懂:我们常说的机器人操作系统,到底是什么?在机器人领域,“操作系统” 从来不是单一概念,而是一套功能分层、各司其职的完整软件体系。不同层级定位不同、职责分明,实际项目中可组合部署、按需协作&…...
)
MathType 快捷键实战指南——数学建模效率飙升的秘诀(从入门到精通)
1. 为什么你需要掌握MathType快捷键? 如果你经常需要处理数学公式,肯定遇到过这样的场景:为了输入一个简单的积分符号,不得不从工具栏里翻找半天;调整公式对齐时反复用鼠标拖动;修改矩阵维度时逐个单元格调…...

自动化测试系统开关架构与继电器选型指南
1. 自动化测试系统中的开关架构选择在自动化测试系统中,开关架构的选择直接影响着测试效率、信号完整性和系统成本。根据测试需求和被测设备(DUT)特性,我们可以将开关架构分为四种基本类型。1.1 无开关架构无开关架构是最直接的连接方式,每个…...

如何为iOS 14.0-16.6.1设备安装TrollStore:TrollInstallerX完整指南
如何为iOS 14.0-16.6.1设备安装TrollStore:TrollInstallerX完整指南 【免费下载链接】TrollInstallerX A TrollStore installer for iOS 14.0 - 16.6.1 项目地址: https://gitcode.com/gh_mirrors/tr/TrollInstallerX 如果你正在寻找一种可靠且简单的方法在i…...

终极指南:SpringAll安全框架实战——Shiro与Spring Security权限控制最佳实践
终极指南:SpringAll安全框架实战——Shiro与Spring Security权限控制最佳实践 【免费下载链接】SpringAll 循序渐进,学习Spring Boot、Spring Boot & Shiro、Spring Batch、Spring Cloud、Spring Cloud Alibaba、Spring Security & Spring Secur…...

3分钟学会离线语音转文字:TMSpeech让你的会议记录不再遗漏
3分钟学会离线语音转文字:TMSpeech让你的会议记录不再遗漏 【免费下载链接】TMSpeech 腾讯会议摸鱼工具 项目地址: https://gitcode.com/gh_mirrors/tm/TMSpeech 你是否经常因为会议内容太多记不住而焦虑?是否担心网络语音识别会泄露你的隐私&…...