fastp软件介绍
fastp软件介绍
- 1、软件介绍
- 2、重要参数解析
- 2.1 全部参数
- 2.2 使用示例
- 2.3 重要参数详解
- (1)UMI去除
- (2)质量过滤
- (3)长度过滤
- (4)低复杂度过滤
- (5)adapter过滤
- (6)通过质量值过滤每条read
- (7)ployG/ployX
- (8)PE数据的碱基校正(correction)
- (9)整体切除 【global trimming】
- 过滤reads顺序
- (10)输出文件切分
- (11)过表达序列分析 【overrepresented sequence analysis】
- 3、软件质控结果文件部分说明
- 3.1 Summary(整体结果)
- 3.2 Adapter
- 3.3 Insert size estimation
- 3.4 Before filtering
- 4、参考文件
1、软件介绍
在18年之前,fq质控用FASTQC等软件,去接头序列用cutadapt软件,数据过滤用Trimmomatic等软件。
后来海普洛斯的CTO开发了一款新软件fastp,整合了上述3个功能,实现了又快又好又个性化。
2、重要参数解析
2.1 全部参数
fastp: an ultra-fast all-in-one FASTQ preprocessor
version 0.19.5
usage: fastp [options] ...
options:-i, --in1 read1 input file name (string [=])-o, --out1 read1 output file name (string [=])-I, --in2 read2 input file name (string [=])-O, --out2 read2 output file name (string [=])-6, --phred64 indicate the input is using phred64 scoring (it'll be converted to phred33, so the output will still be phred33)-z, --compression compression level for gzip output (1 ~ 9). 1 is fastest, 9 is smallest, default is 4. (int [=4])--stdin input from STDIN. If the STDIN is interleaved paired-end FASTQ, please also add --interleaved_in.--stdout stream passing-filters reads to STDOUT. This option will result in interleaved FASTQ output for paired-end input. Disabled by defaut.--interleaved_in indicate that <in1> is an interleaved FASTQ which contains both read1 and read2. Disabled by defaut.--reads_to_process specify how many reads/pairs to be processed. Default 0 means process all reads. (int [=0])--dont_overwrite don't overwrite existing files. Overwritting is allowed by default.-V, --verbose output verbose log information (i.e. when every 1M reads are processed).-A, --disable_adapter_trimming adapter trimming is enabled by default. If this option is specified, adapter trimming is disabled-a, --adapter_sequence the adapter for read1. For SE data, if not specified, the adapter will be auto-detected. For PE data, this is used if R1/R2 are found not overlapped. (string [=auto])--adapter_sequence_r2 the adapter for read2 (PE data only). This is used if R1/R2 are found not overlapped. If not specified, it will be the same as <adapter_sequence> (string [=auto])--detect_adapter_for_pe by default, the auto-detection for adapter is for SE data input only, turn on this option to enable it for PE data.-f, --trim_front1 trimming how many bases in front for read1, default is 0 (int [=0])-t, --trim_tail1 trimming how many bases in tail for read1, default is 0 (int [=0])-b, --max_len1 if read1 is longer than max_len1, then trim read1 at its tail to make it as long as max_len1. Default 0 means no limitation (int [=0])-F, --trim_front2 trimming how many bases in front for read2. If it's not specified, it will follow read1's settings (int [=0])-T, --trim_tail2 trimming how many bases in tail for read2. If it's not specified, it will follow read1's settings (int [=0])-B, --max_len2 if read2 is longer than max_len2, then trim read2 at its tail to make it as long as max_len2. Default 0 means no limitation. If it's not specified, it will follow read1's settings (int [=0])-g, --trim_poly_g force polyG tail trimming, by default trimming is automatically enabled for Illumina NextSeq/NovaSeq data--poly_g_min_len the minimum length to detect polyG in the read tail. 10 by default. (int [=10])-G, --disable_trim_poly_g disable polyG tail trimming, by default trimming is automatically enabled for Illumina NextSeq/NovaSeq data-x, --trim_poly_x enable polyX trimming in 3' ends.--poly_x_min_len the minimum length to detect polyX in the read tail. 10 by default. (int [=10])-5, --cut_by_quality5 enable per read cutting by quality in front (5'), default is disabled (WARNING: this will interfere deduplication for both PE/SE data)-3, --cut_by_quality3 enable per read cutting by quality in tail (3'), default is disabled (WARNING: this will interfere deduplication for SE data)-W, --cut_window_size the size of the sliding window for sliding window trimming, default is 4 (int [=4])-M, --cut_mean_quality the bases in the sliding window with mean quality below cutting_quality will be cut, default is Q20 (int [=20])-Q, --disable_quality_filtering quality filtering is enabled by default. If this option is specified, quality filtering is disabled-q, --qualified_quality_phred the quality value that a base is qualified. Default 15 means phred quality >=Q15 is qualified. (int [=15])-u, --unqualified_percent_limit how many percents of bases are allowed to be unqualified (0~100). Default 40 means 40% (int [=40])-n, --n_base_limit if one read's number of N base is >n_base_limit, then this read/pair is discarded. Default is 5 (int [=5])-L, --disable_length_filtering length filtering is enabled by default. If this option is specified, length filtering is disabled-l, --length_required reads shorter than length_required will be discarded, default is 15. (int [=15])--length_limit reads longer than length_limit will be discarded, default 0 means no limitation. (int [=0])-y, --low_complexity_filter enable low complexity filter. The complexity is defined as the percentage of base that is different from its next base (base[i] != base[i+1]).-Y, --complexity_threshold the threshold for low complexity filter (0~100). Default is 30, which means 30% complexity is required. (int [=30])--filter_by_index1 specify a file contains a list of barcodes of index1 to be filtered out, one barcode per line (string [=])--filter_by_index2 specify a file contains a list of barcodes of index2 to be filtered out, one barcode per line (string [=])--filter_by_index_threshold the allowed difference of index barcode for index filtering, default 0 means completely identical. (int [=0])-c, --correction enable base correction in overlapped regions (only for PE data), default is disabled--overlap_len_require the minimum length of the overlapped region for overlap analysis based adapter trimming and correction. 30 by default. (int [=30])--overlap_diff_limit the maximum difference of the overlapped region for overlap analysis based adapter trimming and correction. 5 by default. (int [=5])-U, --umi enable unique molecular identifer (UMI) preprocessing--umi_loc specify the location of UMI, can be (index1/index2/read1/read2/per_index/per_read, default is none (string [=])--umi_len if the UMI is in read1/read2, its length should be provided (int [=0])--umi_prefix if specified, an underline will be used to connect prefix and UMI (i.e. prefix=UMI, UMI=AATTCG, final=UMI_AATTCG). No prefix by default (string [=])--umi_skip if the UMI is in read1/read2, fastp can skip several bases following UMI, default is 0 (int [=0])-p, --overrepresentation_analysis enable overrepresented sequence analysis.-P, --overrepresentation_sampling one in (--overrepresentation_sampling) reads will be computed for overrepresentation analysis (1~10000), smaller is slower, default is 20. (int [=20])-j, --json the json format report file name (string [=fastp.json])-h, --html the html format report file name (string [=fastp.html])-R, --report_title should be quoted with ' or ", default is "fastp report" (string [=fastp report])-w, --thread worker thread number, default is 2 (int [=2])-s, --split split output by limiting total split file number with this option (2~999), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (int [=0])-S, --split_by_lines split output by limiting lines of each file with this option(>=1000), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (long [=0])-d, --split_prefix_digits the digits for the sequential number padding (1~10), default is 4, so the filename will be padded as 0001.xxx, 0 to disable padding (int [=4])-?, --help print this message
简书上有人以功能划分,重新归纳了参数如下,更加方便查看:
usage: fastp -i <in1> -o <out1> [-I <in1> -O <out2>] [options...]
options:# I/O options 即输入输出文件设置-i, --in1 read1 input file name (string)-o, --out1 read1 output file name (string [=])-I, --in2 read2 input file name (string [=])-O, --out2 read2 output file name (string [=])-6, --phred64 indicates the input is using phred64 scoring (it'll be converted to phred33, so the output will still be phred33)-z, --compression compression level for gzip output (1 ~ 9). 1 is fastest, 9 is smallest, default is 2\. (int [=2])--reads_to_process specify how many reads/pairs to be processed. Default 0 means process all reads. (int [=0])# adapter trimming options 过滤序列接头参数设置-A, --disable_adapter_trimming adapter trimming is enabled by default. If this option is specified, adapter trimming is disabled-a, --adapter_sequence the adapter for read1\. For SE data, if not specified, the adapter will be auto-detected. For PE data, this is used if R1/R2 are found not overlapped. (string [=auto])--adapter_sequence_r2 the adapter for read2 (PE data only). This is used if R1/R2 are found not overlapped. If not specified, it will be the same as <adapter_sequence> (string [=])# global trimming options 剪除序列起始和末端的低质量碱基数量参数-f, --trim_front1 trimming how many bases in front for read1, default is 0 (int [=0])-t, --trim_tail1 trimming how many bases in tail for read1, default is 0 (int [=0])-F, --trim_front2 trimming how many bases in front for read2\. If it's not specified, it will follow read1's settings (int [=0])-T, --trim_tail2 trimming how many bases in tail for read2\. If it's not specified, it will follow read1's settings (int [=0])# polyG tail trimming, useful for NextSeq/NovaSeq data polyG剪裁-g, --trim_poly_g force polyG tail trimming, by default trimming is automatically enabled for Illumina NextSeq/NovaSeq data--poly_g_min_len the minimum length to detect polyG in the read tail. 10 by default. (int [=10])-G, --disable_trim_poly_g disable polyG tail trimming, by default trimming is automatically enabled for Illumina NextSeq/NovaSeq data# polyX tail trimming-x, --trim_poly_x enable polyX trimming in 3' ends.--poly_x_min_len the minimum length to detect polyX in the read tail. 10 by default. (int [=10])# per read cutting by quality options 划窗裁剪-5, --cut_by_quality5 enable per read cutting by quality in front (5'), default is disabled (WARNING: this will interfere deduplication for both PE/SE data)-3, --cut_by_quality3 enable per read cutting by quality in tail (3'), default is disabled (WARNING: this will interfere deduplication for SE data)-W, --cut_window_size the size of the sliding window for sliding window trimming, default is 4 (int [=4])-M, --cut_mean_quality the bases in the sliding window with mean quality below cutting_quality will be cut, default is Q20 (int [=20])# quality filtering options 根据碱基质量来过滤序列-Q, --disable_quality_filtering quality filtering is enabled by default. If this option is specified, quality filtering is disabled-q, --qualified_quality_phred the quality value that a base is qualified. Default 15 means phred quality >=Q15 is qualified. (int [=15])-u, --unqualified_percent_limit how many percents of bases are allowed to be unqualified (0~100). Default 40 means 40% (int [=40])-n, --n_base_limit if one read's number of N base is >n_base_limit, then this read/pair is discarded. Default is 5 (int [=5])# length filtering options 根据序列长度来过滤序列-L, --disable_length_filtering length filtering is enabled by default. If this option is specified, length filtering is disabled-l, --length_required reads shorter than length_required will be discarded, default is 15\. (int [=15])# low complexity filtering-y, --low_complexity_filter enable low complexity filter. The complexity is defined as the percentage of base that is different from its next base (base[i] != base[i+1]).-Y, --complexity_threshold the threshold for low complexity filter (0~100). Default is 30, which means 30% complexity is required. (int [=30])# filter reads with unwanted indexes (to remove possible contamination)--filter_by_index1 specify a file contains a list of barcodes of index1 to be filtered out, one barcode per line (string [=])--filter_by_index2 specify a file contains a list of barcodes of index2 to be filtered out, one barcode per line (string [=])--filter_by_index_threshold the allowed difference of index barcode for index filtering, default 0 means completely identical. (int [=0])# base correction by overlap analysis options 通过overlap来校正碱基-c, --correction enable base correction in overlapped regions (only for PE data), default is disabled# UMI processing-U, --umi enable unique molecular identifer (UMI) preprocessing--umi_loc specify the location of UMI, can be (index1/index2/read1/read2/per_index/per_read, default is none (string [=])--umi_len if the UMI is in read1/read2, its length should be provided (int [=0])--umi_prefix if specified, an underline will be used to connect prefix and UMI (i.e. prefix=UMI, UMI=AATTCG, final=UMI_AATTCG). No prefix by default (string [=])--umi_skip if the UMI is in read1/read2, fastp can skip several bases following UMI, default is 0 (int [=0])# overrepresented sequence analysis-p, --overrepresentation_analysis enable overrepresented sequence analysis.-P, --overrepresentation_sampling One in (--overrepresentation_sampling) reads will be computed for overrepresentation analysis (1~10000), smaller is slower, default is 20\. (int [=20])# reporting options-j, --json the json format report file name (string [=fastp.json])-h, --html the html format report file name (string [=fastp.html])-R, --report_title should be quoted with ' or ", default is "fastp report" (string [=fastp report])# threading options 设置线程数-w, --thread worker thread number, default is 3 (int [=3])# output splitting options-s, --split split output by limiting total split file number with this option (2~999), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (int [=0])-S, --split_by_lines split output by limiting lines of each file with this option(>=1000), a sequential number prefix will be added to output name ( 0001.out.fq, 0002.out.fq...), disabled by default (long [=0])-d, --split_prefix_digits the digits for the sequential number padding (1~10), default is 4, so the filename will be padded as 0001.xxx, 0 to disable padding (int [=4])# help-?, --help print this message2.2 使用示例
(1)最简单的使用示例
fastp -i in.fq -o out.fq # SE测序数据
fastp -i in.R1.fq -o out.R1.fq -I in.R2.fq -O out.R2.fq # PE测序书
fastp -i in.R1.fq.gz -I in.R2.fq.gz -o out.R1.fq.gz -O out.R2.fq.gz # 输入压缩文件,输出也为压缩文件
(2)结合常用参数的使用示例
fastp
-i *_R1_raw.fastq.gz # reads1 fastq
-I *_R2_raw.fastq.gz # reads2 fastq
-o *_R1_trim.fastq.gz # reads1 处理结果
-O *_R2_trim.fastq.gz # reads2 处理结果
--adapter_sequence=AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC # reads1 接头序列
--adapter_sequence_r2=AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT # reads2 接头序列
--thread=4 # 设置线程数
--length_required=55 # 过滤过短序列,自定义55以下为短序列
--compression=4 # 压缩比例,1最快, 9最慢
--trim_poly_g # 开启polyG剪裁, 适用于Illumina NextSeq和NovaSeq系列数据
--cut_by_quality3 # 开启在3’端,也就是read末尾的剪裁
--correction # 通过overlap来校正碱基
--umi # 添加了umi技术的测序数据
--umi_loc per_read # 指定UMI所在的位置, per_read指在每个插入序列中
--umi_len 5 # UMI所占碱基长度
--umi_skip 3 # 去除UMI后,再去除3bp
-j *.trim.fastp.json # 适合程序读的JSON格式质控结果
-h *.trim.fastp.html # 适合人看的网页格式质控结果
2.3 重要参数详解
本部分内容来自参考文件:质控软件fastp常用参数说明_fastp参数_青灯照颦微的博客-CSDN博客;欢迎各位去读原文。
(1)UMI去除
分子标签(UMI),来自于相同的分子的标记,用于去重,错误校正。常用在ctDNA测序,illumina测序的UMI位于两个不同位置:index和read开头。
--umi 启用UMI处理参数;
--umi_loc 指定UMI的位置,可设置下面几种:
"index1": 第一个index作为UMI, 对双端数据,则作用于R1/R2;
"index2": 第二个index作为UMI, 对双端数据,则作用于R1/R2;
"read1": read1的头部作为UMI, 对双端数据,则作用于R1/R2;
"read2": read2的头部作为UMI, 对双端数据,则作用于R1/R2;
"pre_index", "index1_index2":
"pre_read": read1的头部定义'umi1', read2的头部定义'umi2', 'umi1_umi2'作为UMI, 作用于R1/R2
--umi_len UMI的长度,当指定UMI的位置为read1, read2,per_read时,应指定UMI长度;
--umi_prefix UMI设置前缀,例: UMI=AATTCCGG,prefix=ATC,即设置–umi_prefix=ATC,则被加在read_name行的UMI序列将会是ATC_AATTCCGG ;
--umi_skip UMI去除并加到read_name后,再去除(跳过)的碱基数;例:–umi_skip=4 表示去除UMI后再去除4bp。
fastp是将UMI提取后加在对应read的name行,如果UMI在read中,那么UMI会从read中移除,如果UMI在index中,会被保留。
(2)质量过滤
-q, --qualified_quality_phred 设置碱基质量值不小于多少时,该碱基为合格碱基,默认碱基质量值是15,即默认碱基质量>=15是合格碱基,<15为不合格碱基;
-u --unqualified_percent_limit 设置允许不合格碱基的占比为多少时,去掉这条read,默认是40,即默认不合格碱基占比>40%时,去掉该read;
-Q, --disable_quality_filtering 设置该参数则禁用默认质量过滤参数(-q, -u)。
(3)长度过滤
-l, --length_required 设置read的最小长度,默认是15,即长度<15的read被去掉;
--length_limit 设置read的最大长度, 默认为0是没有最大长度限制;
(4)低复杂度过滤
-Y, --complexity_threshold 设置read的复杂度过滤阈值,默认为30,即当read复杂度<30时,去掉该read。复杂度:
- 复杂度的定义为 一个碱基与其下一个相邻碱基不同的碱基个数占比;
- 例:一条长为51bp的read,有3个碱基与其下一个碱基不同seq = 'AAAATTTTTTTTTTTTTTTTTTTTTGGGGGGGGGGGGGGGGGGGGGGCCCC'其复杂度为:complexity = 3/(51-1) = 6%
-y, --low_complexity_filter 设置该参数则禁用默认复杂度过滤参数(-Y)
(5)adapter过滤
-A, --disable_adapter_trimming 设置该参数则禁用默认adapter过滤参数;
-a, --adapter_sequence 指定引物序列(对应SE数据的引物序列 或 对应PE数据的R1的引物序列)。对单端(SE)数据,可通过自动检测前~1Mreads的尾巴,去识别adapter,若设置该参数,则表示禁用自动识别adapter;
--adapter_sequence_r2 指定R2引物序列(对PE数据的R2)。对双端(PE)数据,是通过两条reads的overlap去adapter(由于该方法比较稳定,通常不必设置引物序列)。如果为找到overlap,用使用这些序列去adapter(是否设置都先通过overlap去adapter?);
--detect_adapter_for_pe 默认对双端数据则默认不使用自动检测adapter(SE可自动检测),设置该参数,表示对双端数据也启用自动检测方法;
--adapter_fasta 接头序列文件(fasta格式),注意该fasta文件中的fasta序列长度至少6bp,否则会被跳过。
注:fastp首先去除自动化检测到的接头序列,或者使用–adapter_sequence |–adapter_sequence_r2指定的接头序列,然后去除由–adapter_fasta设置的接头序列。去除的接头序列分布可以在html/json文件中查看。
(6)通过质量值过滤每条read
下面参数是通过滑动窗的平均质量值切除reads。
-W, --cut_window_size 设置滑动窗口大小;
-M, --cut_mean_quality 设置滑动窗口的平均质量值阈值,低于这个阈值则被切除;
可对两端分别进行切除:
对5'端的参数,与Trimmomatic中的LEADING参数方法相似:-5, --cut_front 是去除5'端低质量碱基,具体是指滑动窗从5'向末尾3’滑动,如果窗口内的碱基平均质量值低于阈值,则切除这些碱基,然后窗口继续滑动,直到达到阈值则不再去除;--cut_front_window_size 是设置从5'端开始的滑动窗的大小,即每个滑动窗包含几个碱基;--cut_front_mean_quality 设置从5'端开始的滑动窗平均质量值阈值,低于该阈值则切除这些碱基;
对3'端开始切除的参数与5'端类似,也与Trimmomatic中的TRAILING参数的方法类似:-3, --cut_tail 是去除3'端低质量碱基,具体是指滑动窗从3'向起始5’滑动,如果窗口内的碱基平均质量值低于阈值,则切除这些碱基,然后窗口继续滑动,直到达到阈值则不再去除;--cut_tail_window_size 是设置从3'端开始的滑动窗的大小;--cut_tail_mean_quality 设置从3'端开始的滑动窗平均质量值阈值,低于该阈值则切除这些碱基;
还有切除序列的其他参数:
-r, --cut_right 是切除右侧序列,-3与-r参数的差别是,前者是先进行碱基去除,达到阈值则不再去除碱基,然后继续滑动窗口;后者是前者进行后,继续滑动滑动窗,直到发现窗口内碱基的平均质量值低于阈值,则切除该窗口及右侧所有碱基。也就是使用该参数,就没必要设置–cut_tail参数 。
(7)ployG/ployX
对Illumina的NextSeq/NovaSeq测序数据,常会用ployG发生(是因为这两个平台使用两个荧光信号,而没有信号时表示G)。fastp能够检测到ployG并去除(默认是NextSeq/NovaSeq平台,通过测序仪ID和fastq识别)
-g, --trim_poly_g 启用去除尾巴ployG;
--poly_g_min_len 设置去除尾巴’G’的最小长度,默认为10即尾巴ployG长度>10时,会被去除;
-G, --disable_trim_poly_g 禁用去除尾巴ployG;
-x, --polyX 启用去除polyX(polyA, polyT, polyC, polyG),若同时设置–trim_poly_g和–ployX,则先进行ployG尾巴去重,再进行ployX(这样设置有助于ployA尾巴在G尾巴之前时,去重ployA尾巴[常见于mRNA-Seq])。
(8)PE数据的碱基校正(correction)
fastp通过overlap进行分析,如果找到合适的overlap,当overlap区域的两个错配碱基中,一个碱基质量值较高,一个碱基质量值极低,该软件会将错配的两个碱基进行校正(?将低质量碱基校正为与高质量碱基互补的碱基)。对应的碱基质量值也校正为相同的值。
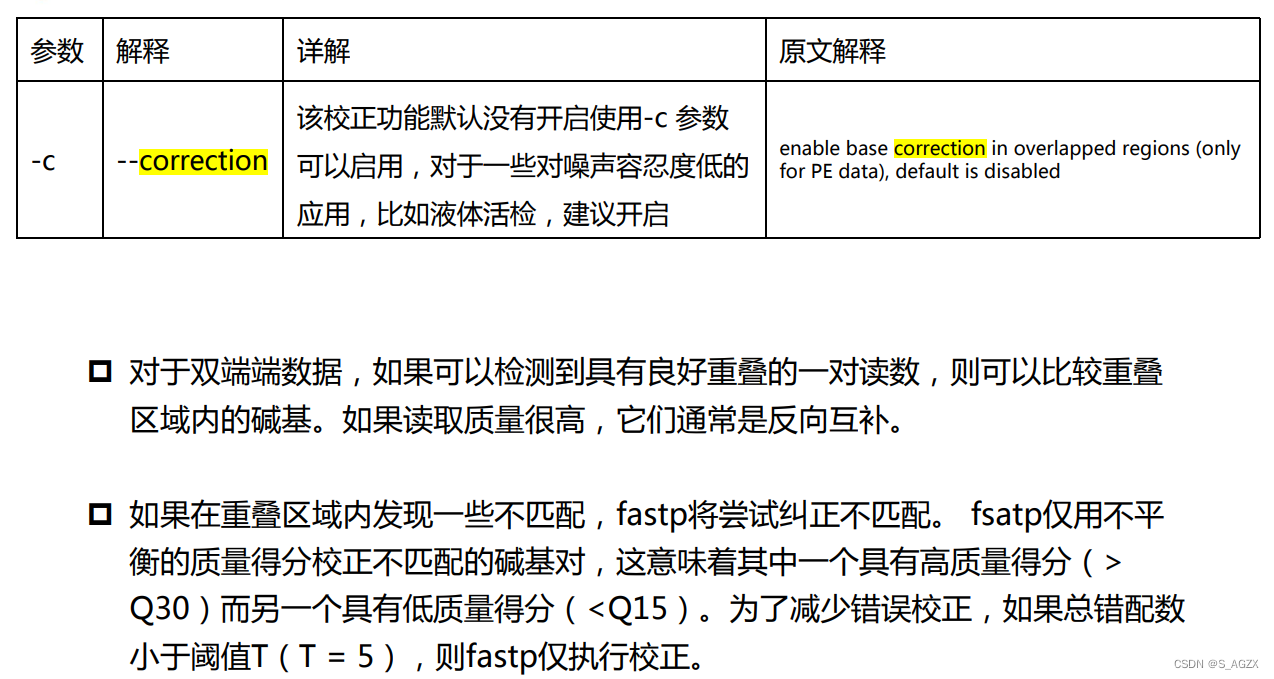
-c, --correction 对碱基校正,默认不启用该参数;使用该参数是基于检测overlap,overlap的可调参数有:
--overlap_len_require overlap的长度要求,默认是30,即默认overlap区域的长度不低于30bp;否则认为无overlap;--overlap_diff_limit overlap中最大错配数,默认是5,即默认overlap时最多有5个错配;否则认为无overlap;--overlap_diff_percent_limit overlap中最大错配数在重叠区的占比,默认是20,即默认最大错配数的碱基占比不高于20%;否则认为无overlap。

(9)整体切除 【global trimming】
整体切除一般是考虑到,illumina测序最后1个cycle或最后n个cycle测序质量较低,使用-t 1, --trim_tai1l=1参数将所有reads的末尾1bp去除;
-f, --trim_front1 对R1起始几bp进行去除,例如:-f 1或–trim_front1=1表示去除R1起始位置1bp碱基;
-t, --trim_tail1 对R1末尾几bp进行去除,例如:-t 2或–trim_tail1=2表示去除R1末尾位置1bp碱基;
-b, --max_len1 设置R1最大长度阈值,即R1的长度大于阈值,则在尾巴开始切除read直到与阈值相等,默认不切除。注意最大长度在最后一步处理;
-F, --trim_front2 与R1相似;不设置默认则与R1指定的参数相同;
-T, --trim_tail2 与R1相似;不设置默认则与R1指定的参数相同;
-B, --max_len2 设置R2最大长度,同-b参数。[注意最大长度在最后一步处理]
过滤reads顺序
## 过滤reads顺序:
1. 对UMI进行处理("--umi")
2. 整体切除的起始位置切除("-f", "-F") # 比如UMI在5‘端却不知道序列时,可trim_front1 10 trim_front2 10来强制去除插入序列
3. 整体切除的尾巴位置切除("-t", "-T")
4. 5'端质量值切除("-cut_front")
5. 滑动窗切除("--cut_right")
6. 3'端质量值切除("--cut_tail")
7. ployG切除("--trim_ploy_g", 默认作用于'NovaSeq/NextSeq'的数据)
8. 根据overlap分析去adapter(PE数据)
9. 根据adapter序列去apapter("--adapter_sequence", "--adapter_sequence_r2", 对PE数据则跳过该步骤)
10. 去除polyX("--trim_poly_x")
11. 去除最大长度("--max_len")
(10)输出文件切分
可通过设置分割成几个文件或者设置每个文件的行数 ,两者不可同时设置。
-s, --split 指定最多分割成几个文件;
-S, --split_by_lines 指定分割后的每个文件最多几行;
-d, --split_prefix_digits 设置输出文件的前缀数字位数,例如:–split_prefix_digits=4 --split=3 --out1=out.fq , 则输出文件为0001.out.fq, 0002.out.fq, 0002.out.fq

(11)过表达序列分析 【overrepresented sequence analysis】
-p,--overrepresentation_analysis 启用该分析,默认仅统计序列长度为10bp, 20bp, 40bp, 100bp或 cycle -2 ;
-P, --overrepresentation_sampling 指定用于统计的reads数比例,默认20,即默认1/20的reads用于序列统计。例:设置-P 100 表示将1/100的reads用序列统计,设置-P 1 表示将所有reads用于统计(运行会很慢,默认20是平衡了速度和精确度)
不仅有过表达序统计结果,还有循环中(cycles)的分布情况,并用图展示检测到的过表达序列,以便找到最多的序列。

3、软件质控结果文件部分说明
本部分结果,主要来自于博客生信学习笔记:fastp质控处理生成的report结果解读_fastp结果解读_twocanis的博客-CSDN博客,欢迎去读原文。
3.1 Summary(整体结果)
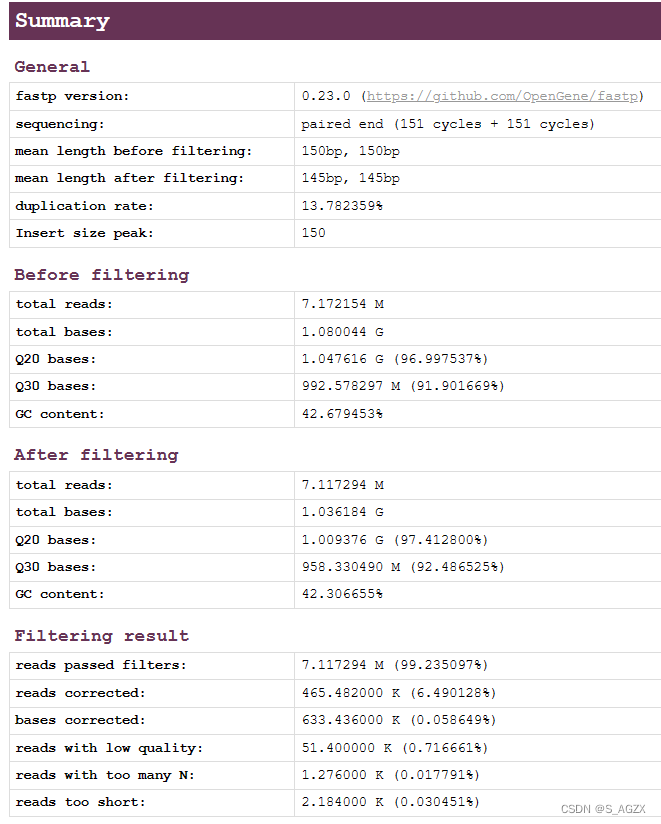
General
版本号、序列循环数、质控之前的平均长度、质控之后的平均长度、插入片段的峰值
Before filtering
数据质控之前的(反应测序质量):总的reads长度、总碱基长度、Q20合格率、Q30合格率、GC含量
After filtering
质控之后的:内容同上
Filtering result
reads的通过率、低质量的reads、含太多N值的reads
3.2 Adapter

3.3 Insert size estimation

配对末端重叠分析,不同长度的Insert在reads中占的比例,相当于是DNA被打断后的长度分布。当插入片段大小<30或> 270,或包含太多错误,则不能被read读取,比如我这里就有28%的不可读reads)
3.4 Before filtering
质控之前的数据质量、碱基含量以及kmer分析等,可直接在网页上用鼠标拖动放大缩小以及查看具体数据细节,或进行图片保存等操作
(1)reads质量
在不同位置上的碱基质量分布,一般来讲质量应 >30 且波动较小为不错的数据

(2)碱基质量
read各个位置上碱基比例分布,这个是为了分析碱基的分离程度。
何为碱基分离?已知AT配对,CG配对,假如测序过程是比较随机的话(随机意味着好),那么在每个位置上A和T比例应该差不多,C和G的比例也应该差不多。
如下图所示,两者之间即使有偏差也不应该太大,最好平均在1%以内,如果过高,除非有合理的原因,比如某些特定的捕获测序所致,否则都需要注意是不是测序过程有什么偏差。

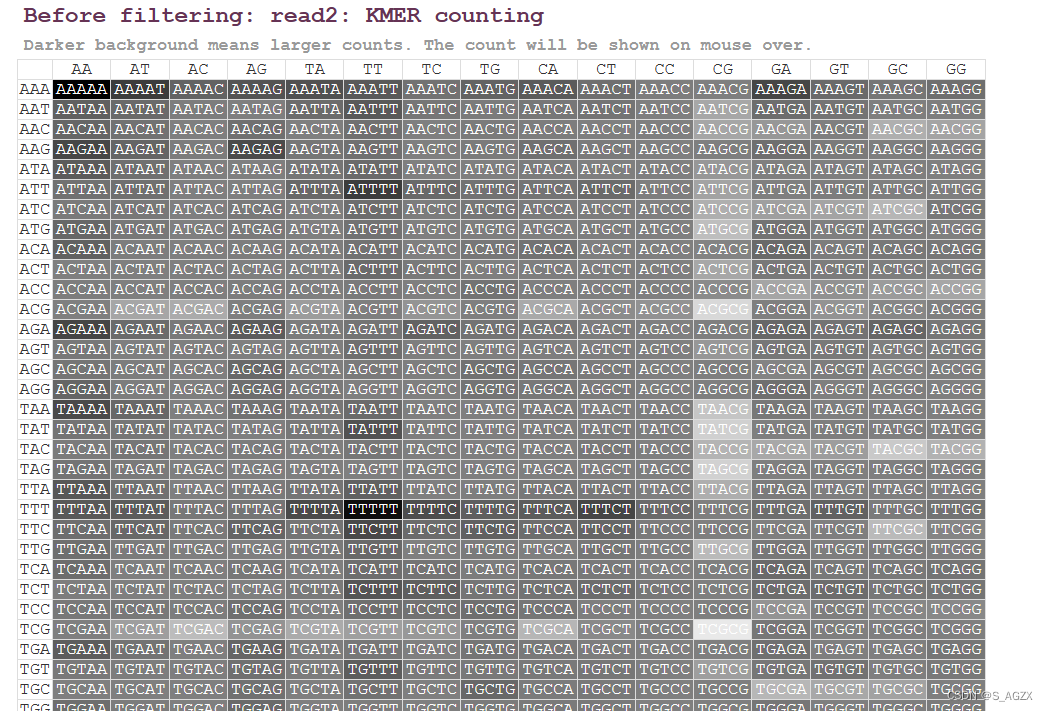
(3)KMER计数
fastp对5个碱基长度的所有组合的出现次数进行了统计,然后把它放在了一张表格中,表格的每一个元素为深背景白字,背景越深,则表示重复次数越多。这样,一眼望去,就可以发现有哪些异常的信息。鼠标可停留在某一具体组合上看出现次数和平均占比。
4、参考文件
1、GitHub - fastp: An ultra-fast all-in-one FASTQ preprocessor
2、fastp参数说明
3、fastp: 一款超快速全功能的FASTQ文件自动化质控+过滤+校正+预处理软件 - 知乎
4、质控软件fastp常用参数说明_fastp参数_青灯照颦微的博客-CSDN博客
5、生信学习笔记:fastp质控处理生成的report结果解读_fastp结果解读_twocanis的博客-CSDN博客
相关文章:
fastp软件介绍
fastp软件介绍1、软件介绍2、重要参数解析2.1 全部参数2.2 使用示例2.3 重要参数详解(1)UMI去除(2)质量过滤(3)长度过滤(4)低复杂度过滤(5)adapter过滤&#…...
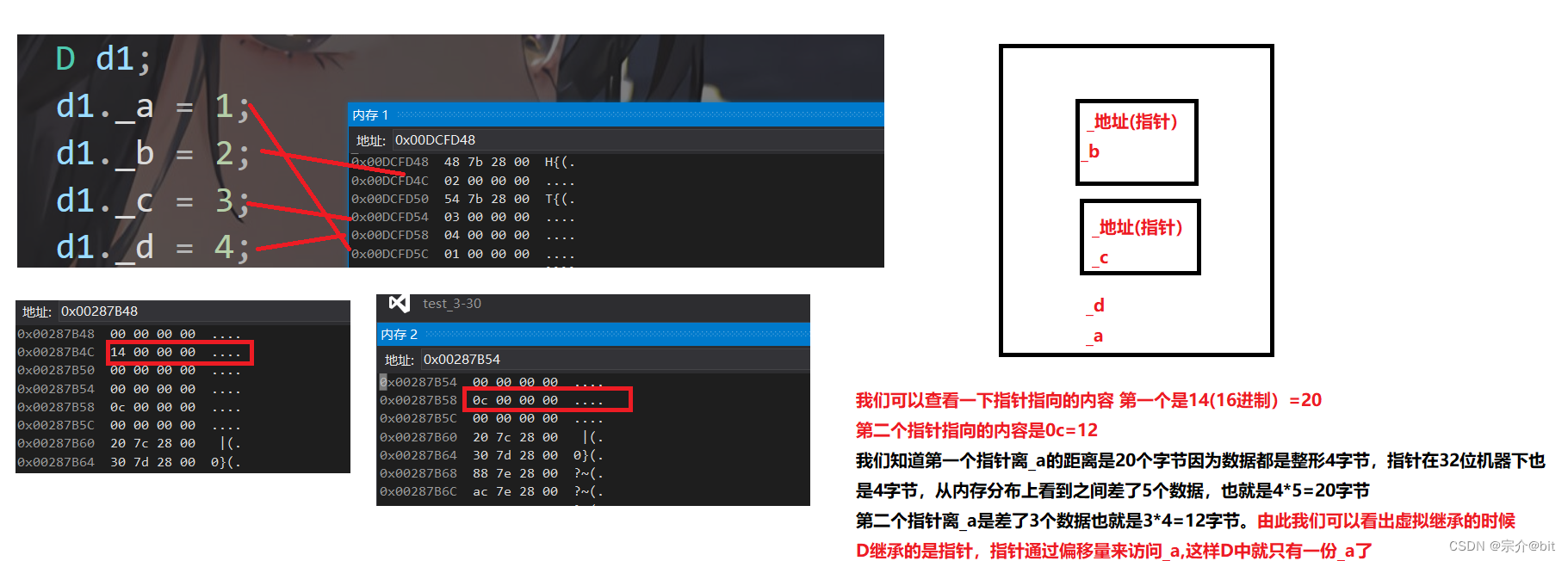
C++继承相关总结
文章目录前言1.继承的相关概念1.继承概念2.继承的相关语法3.基类和派生类对象赋值转换(赋值兼容规则)2.继承中的注意事项1.继承中的作用域2.派生类的默认成员函数1.构造函数与拷贝构造2.赋值重载与析构3.友元关系与静态成员变量3.多继承(菱形继承)1.虚拟继承2.虚拟继…...

【从零开始学习 UVM】8.2、Reporting Infrastructure —— uvm_printer 详解
文章目录 老派风格在UVM中如何完成uvm 风格Table printerTree printerLine printerprint使用print使用条件使用konb更改print配置示例在一个随机验证环境中,数据对象不断地由不同的组件生成和操作,如果能够显示对象的内容,则调试会变得更加容易。 老派风格 传统上,这是通…...

Mybatis、TKMybatis对比
文章目录1.Mybatis(1)配置文件(2)实体类(3)Mapper(4)mybatis-config.xml2.TKMybatis(1)配置文件(2)实体类(3)M…...
37了解高可用技术方案,如冗余、容灾
高可用性技术方案是指在系统设计和架构中采用一系列措施来确保系统在遇到各种故障和问题时仍能保持持续的可用性,避免因单点故障而导致系统宕机、数据丢失等问题。其中包括冗余和容灾技术。 冗余技术: 冗余技术是指通过增加系统组件的冗余来提高系统可靠…...

jdb调试问题集锦
https://bbs.kanxue.com/thread-210049.htm蓝铁 1 2017-8-25 19:40 4 楼 0 根据提示,可知,出错的地方是,android.app.ActivityThread.handleBindApplication(), 行4,400 查看源码可以发现,代码中指向的是app.onCreate() …...
要和文心一言来一把你画我猜吗?
想和文心一言来一把你画我猜吗? ChatGPT的爆火,让AI对话模型再次走入大众视野。大家在感叹ChatGPT的智能程度时,总会忍不住想:如果我们也有自己的AI对话模型就好了。在社会的压力下,国内的厂商和研究机构也纷纷做出尝试…...
有什么区别?)
delete[] p->elems和free(p->elems)有什么区别?
delete[]和free()都是释放内存的函数,但它们具有不同的使用方法和适用情况。 delete[] 通常用于释放C中动态分配的数组空间。在使用new[]运算符分配内存时,应使用delete[]运算符来释放分配的内存。delete[] 运算符会调用每个数组元素的析构函数…...
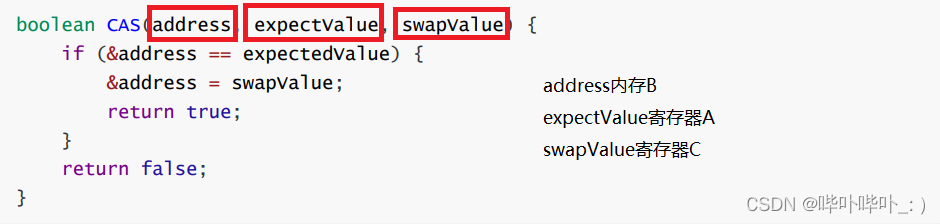
CAS问题
CAS🔎什么是CAS🔎伪代码解析🔎CAS是如何实现原子性的🔎CAS的应用🌻实现原子类🌻实现自旋锁🔎ABA问题🌻ABA问题可能引起的BUG🌻ABA问题的解决方案🔎结尾&#…...
网络编程socket(下)
目录 一、TCP网络程序 1.1 服务端初始化 1.1.1 创建套接字 1.1.2 服务端绑定 1.1.3 服务端监听 1.2 服务端启动 1.2.1 服务端获取连接 1.2.2 服务端处理请求 1.3 客户端初始化 1.4 客户端启动 1.4.1 发起连接 1.4.2 发起请求 1.5 网络测试 1.6 单执行流服务端的…...

华为OD机试题【打折买水果】用 C++ 编码,速通
最近更新的博客 华为od 2023 | 什么是华为od,od 薪资待遇,od机试题清单华为OD机试真题大全,用 Python 解华为机试题 | 机试宝典【华为OD机试】全流程解析+经验分享,题型分享,防作弊指南华为od机试,独家整理 已参加机试人员的实战技巧本篇题解:打折买水果 题目 有 m m m…...

JSON 数据类型
JSON 数据类型 JSON 格式支持以下数据类型: 类型描述数字型(Number)JavaScript 中的双精度浮点型格式字符串型(String)双引号包裹的 Unicode 字符和反斜杠转义字符布尔型(Boolean)true 或 fal…...

Python函数简介
Python是一种高级编程语言,它的函数是其中一个非常重要的特性。在程序中,函数是一段被命名的代码块,它可以接受输入并且返回输出。本篇文章将介绍Python函数的基本概念、定义、调用和参数。 基本概念 在Python中,函数是由def关键…...
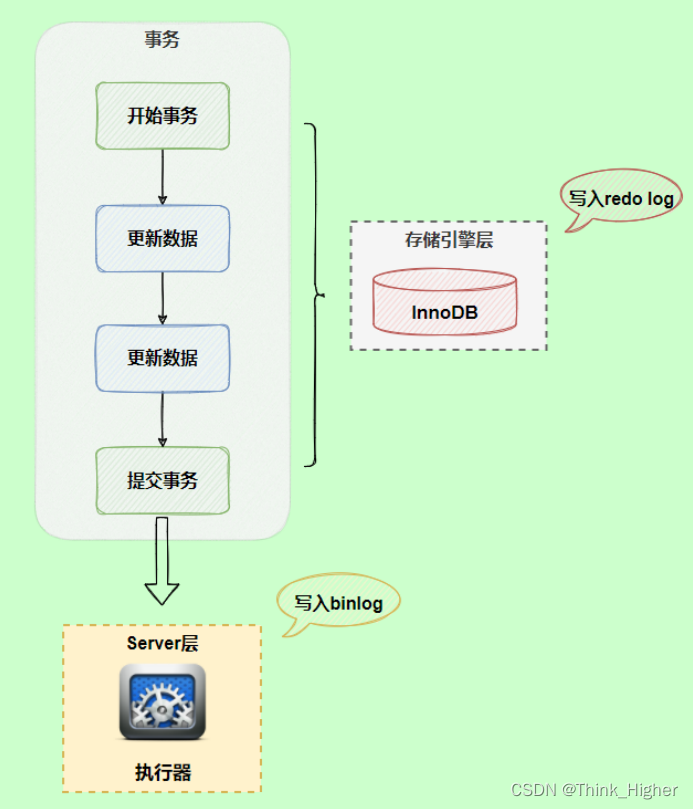
一文读懂 mysql 为什么要两阶段提交以及两阶段提交原理
文章目录 为什么要两阶段提交redo log与binlog两份日志之间的逻辑不一致,会出现什么问题?两阶段提交是怎么保证逻辑一致的呢?当 binlog 写完,redo log 还没 commit 前发生 crash,那崩溃恢复后 MySQL 如何处理?redo 与 binlog 的刷盘时机MySQL 的双 1 配置能否只用 redo l…...
启动Hadoop报错【Error: JAVA_HOME is not set and could not be found.】
当用了一下午从0安装上Hadoop兴奋的启动的时候! Error: JAVA_HOME is not set and could not be found. 他告诉我JAVA_HOME 没被找到? 我明明安装了java的,为什么找不到? java -version看了下发现是没问题的 解决: 后…...

《MySQL系列-InnoDB引擎35》索引与算法-B+树索引的使用
B树索引的使用 1 不同应用中B树索引的使用 在OLTP中,B树索引建立后,对该索引的使用应该只是通过该索引取得表中少部分的数据。这时建立B树索引才是有意义的,否则即使建立了,优化器也可能不选择使用索引。 在OLAP中,…...
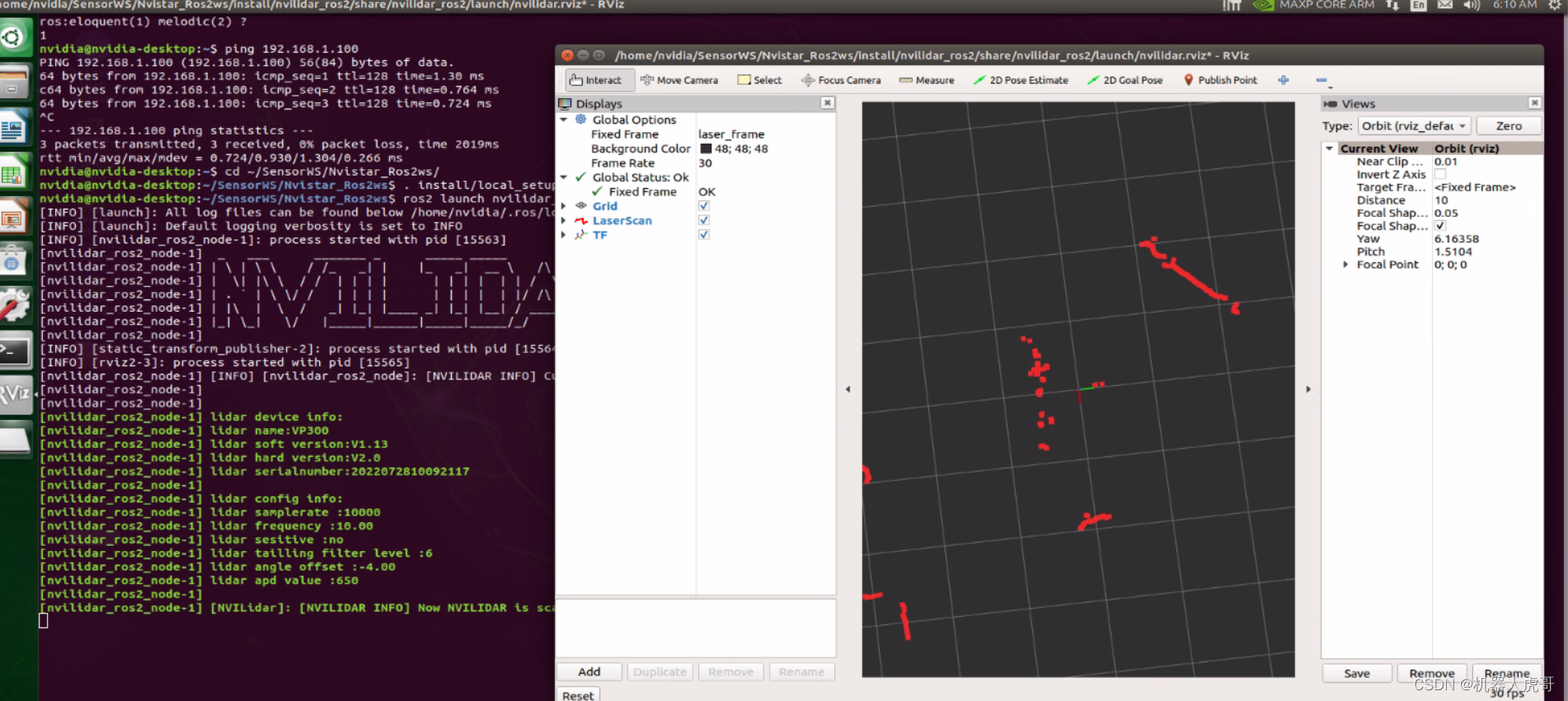
【EHub_tx1_tx2_E100】不止科技NVISTAR ROC 300激光雷达Ubuntu18.04+ROS1ROS2 评测
介绍NVISTAR 的二维DTOF激光雷达 ROC 300在EHub_tx1_tx2_E100载板,TX1核心模块环境(Ubuntu18.04)下测试ROS1驱动和ROS2的驱动,打开使用RVIZ 查看点云数据,本文的前提条件是你的TX1里已经安装了ROS1版本:Mel…...
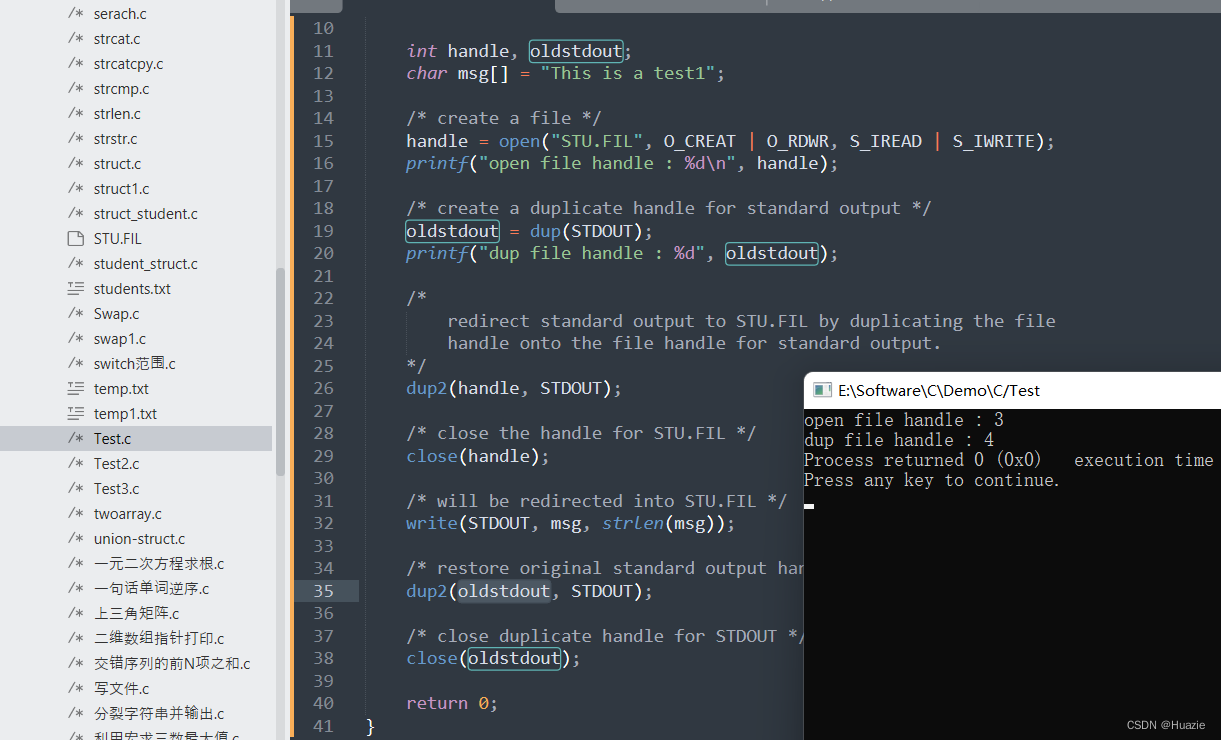
C语言函数大全--d开头的函数
C语言函数大全 本篇介绍C语言函数大全–d开头的函数 1. detectgraph 1.1 函数说明 函数声明函数功能void detectgraph(int *graphdriver, int *graphmode);通过检测硬件确定图形驱动程序和模式 1.2 演示示例 #include <graphics.h> #include <stdlib.h> #incl…...
基于springboot实现福聚苑社区团购演示【项目源码】
基于springboot实现福聚苑社区团购演示开发语言:Java 框架:springboot JDK版本:JDK1.8 服务器:tomcat7 数据库:mysql 5.7 数据库工具:Navicat11 开发软件:eclipse/myeclipse/idea Maven包&#…...
动静态库的制作
文章目录:什么是程序库?动态链接和静态链接动静态库的认识静态库的创建与使用创建使用动态库的创建与使用创建使用什么是程序库? 程序库:一般是软件作者为了发布方便、替换方便或二次开发目的,而发布的一组可以单独与应…...

从内存视角拆解float和double:用C语言和调试器带你‘看见’IEEE754的二进制世界
从内存视角拆解float和double:用C语言和调试器带你‘看见’IEEE754的二进制世界 在计算机科学中,浮点数的表示和处理是一个既基础又关键的话题。对于从事系统编程、性能优化或逆向工程的开发者来说,理解浮点数在内存中的实际存储形式不仅能帮…...

终极macOS清理神器:Pearcleaner 3步彻底卸载应用不留痕迹
终极macOS清理神器:Pearcleaner 3步彻底卸载应用不留痕迹 【免费下载链接】Pearcleaner A free, source-available and fair-code licensed mac app cleaner 项目地址: https://gitcode.com/gh_mirrors/pe/Pearcleaner 你是否曾将macOS应用拖入废纸篓后&…...

当Windows 11 LTSC失去应用商店时,如何轻松找回完整的应用生态?
当Windows 11 LTSC失去应用商店时,如何轻松找回完整的应用生态? 【免费下载链接】LTSC-Add-MicrosoftStore Add Windows Store to Windows 11 24H2 LTSC 项目地址: https://gitcode.com/gh_mirrors/ltscad/LTSC-Add-MicrosoftStore 你是否曾经为W…...

3个高效方法:免费获取百度网盘高速下载直链的完整指南
3个高效方法:免费获取百度网盘高速下载直链的完整指南 【免费下载链接】baidu-wangpan-parse 获取百度网盘分享文件的下载地址 项目地址: https://gitcode.com/gh_mirrors/ba/baidu-wangpan-parse 当我们面对百度网盘缓慢的下载速度时,常常感到无…...
)
Tea印相失效诊断清单:从--v 6.2到--v 6.6,6个版本兼容性断点及降级回滚方案(含JSON config快照备份包)
更多请点击: https://intelliparadigm.com 第一章:Tea印相失效诊断清单:从--v 6.2到--v 6.6,6个版本兼容性断点及降级回滚方案(含JSON config快照备份包) Tea印相(TeaYinXiang)在 v…...

Node.js代理池实战:proxy-agents库核心原理与高级应用
1. 项目概述与核心价值最近在折腾一些需要处理大量网络请求的自动化脚本,比如数据采集、API测试或者模拟用户操作,一个绕不开的痛点就是IP被封。单个IP频繁请求,对方服务器很容易就把你拉黑了。这时候,代理池就成了刚需。市面上成…...

基于LLM与RAG构建智能问答系统:架构、实现与优化指南
1. 项目概述:当RAG遇上LLM,构建你的智能知识问答引擎最近在GitHub上看到一个挺有意思的项目,叫“Jenqyang/LLM-Powered-RAG-System”。光看名字,圈内人大概就能猜到个七七八八:这是一个基于大语言模型(LLM&…...

3步掌握ADB驱动安装:Windows平台最简Android连接方案
3步掌握ADB驱动安装:Windows平台最简Android连接方案 【免费下载链接】Latest-adb-fastboot-installer-for-windows A Simple Android Driver installer tool for windows (Always installs the latest version) 项目地址: https://gitcode.com/gh_mirrors/la/Lat…...

Mac玩转老游戏:手把手教你用Wineskin配置RPG Maker游戏所需RTP环境
Mac玩转老游戏:手把手教你用Wineskin配置RPG Maker游戏所需RTP环境 在Mac上重温经典RPG游戏是许多怀旧玩家的梦想,但RPG Maker游戏往往依赖Windows特有的运行时包(RTP),这让Mac用户望而却步。本文将带你深入探索如何利…...
)
开关电源传导EMI超标?手把手教你用π型滤波器搞定(附SCT2450实测数据)
开关电源传导EMI超标?手把手教你用π型滤波器搞定(附SCT2450实测数据) 在电源设计领域,传导EMI超标是工程师们经常遇到的棘手问题。当你的产品在EMC实验室测试失败时,那种挫败感相信每个硬件工程师都深有体会。传导噪声…...