R语言绘制染色体变异位置分布图,RIdeogram包
变异位点染色体分布图
今天分享的内容是通过RIdeogram包绘制染色体位点分布图,并介绍一种展示差异位点的方法。

在遗传学研究中,通过测序等方式获得了基因组上某些位置的基因型信息。 如下表,第一列是变异位点的ID,第二列是染色体,第三列是物理位置,最后两列是两个样品的基因型。
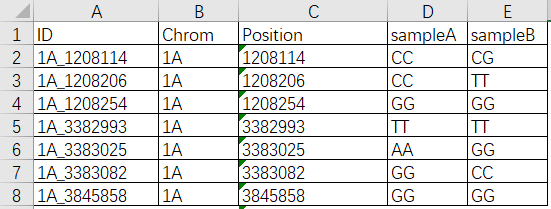
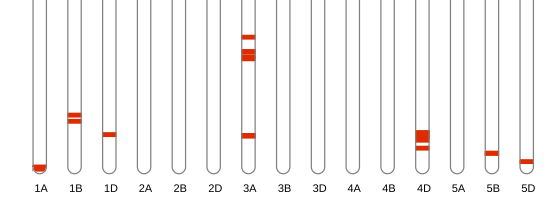
这个文件通常有好几万行,要想快速从中获得有效信息,可以借助变异位点染色体分布图功能实现。比如下图,红色部分表示样品A和样品B差异的位置,一眼就能看出哪些位置比较关键。
数据准备
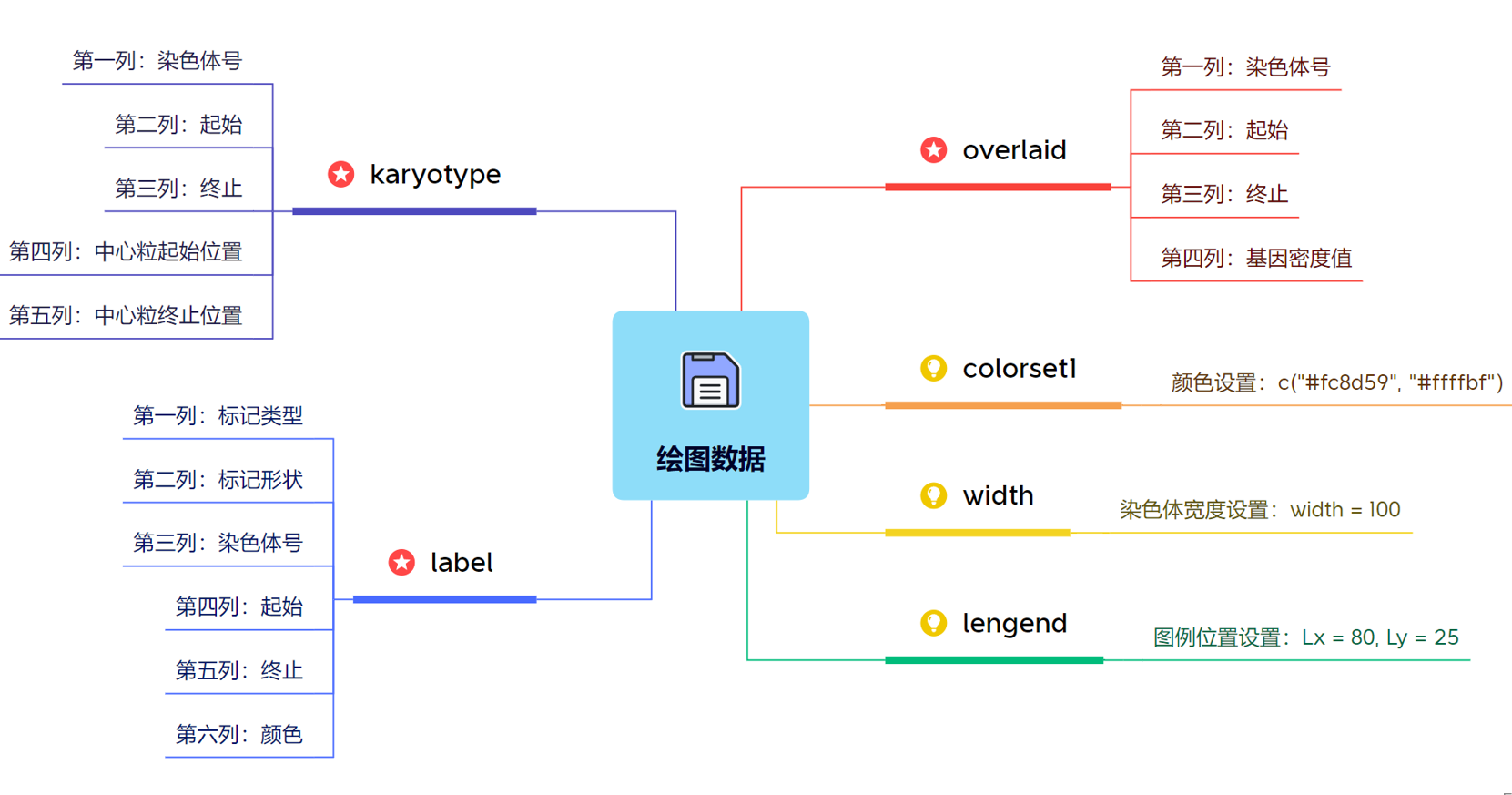
主要的输入文件有两个,第一个是染色体结构信息(长度、着丝粒位置等),第二个是待分析标注的位点信息(不同材料在某位置的基因型),如下所示:
操作方法
首先,安装并加载R包:
library(RIdeogram)
library(tidyverse)
library(xlsx)
设置参数
将整理好的文件保存为csv格式,输入文件名。centwidth是着丝粒显示宽度,singewidth是单个位点的显示宽度,这两个值可以自定义设置。(如果设置的值越大,那么线条就越粗)
my_filename <- "data.csv"
my_centwidth <- 5000000
my_color <- c("#e0e0e0", "#304ffe", "#dd2c00","#00bfa5")
my_singewidth <- 5000000
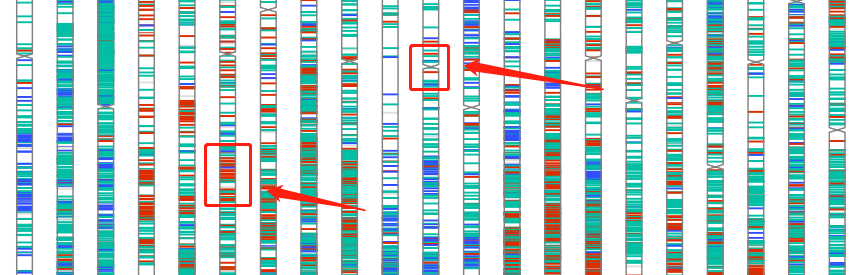
添加染色体信息
由于绘图时需要每个染色体的长度(起始和终止位置)和着丝粒的位置,因此提前准备一个txt文档,按如下格式整理数据,用于后续绘图。 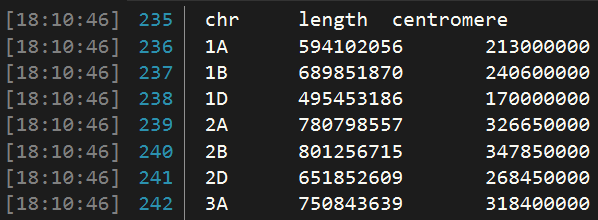
下面是读取染色体信息的代码:
df_chr_pos_cent <- read.table("./Ref_chromedata.txt",header = T)
df_chr_pos_cent <- cbind(df_chr_pos_cent$chr,"0",
df_chr_pos_cent$length,
df_chr_pos_cent$centromere,
as.numeric(df_chr_pos_cent$centromere) + my_centwidth)
df_chr_pos_cent <- as.data.frame(df_chr_pos_cent)
df_chr_pos_cent <- df_chr_pos_cent[1:21,] %>%
mutate_at(vars(V2,V3,V4,V5),as.integer)
colnames(df_chr_pos_cent) <- c("Chr","Start","End","CE_start","CE_end")
> head(df_chr_pos_cent)
Chr Start End CE_start CE_end
1 1A 0 594102056 213000000 218000000
2 1B 0 689851870 240600000 245600000
3 1D 0 495453186 170000000 175000000
4 2A 0 780798557 326650000 331650000
5 2B 0 801256715 347850000 352850000
6 2D 0 651852609 268450000 273450000
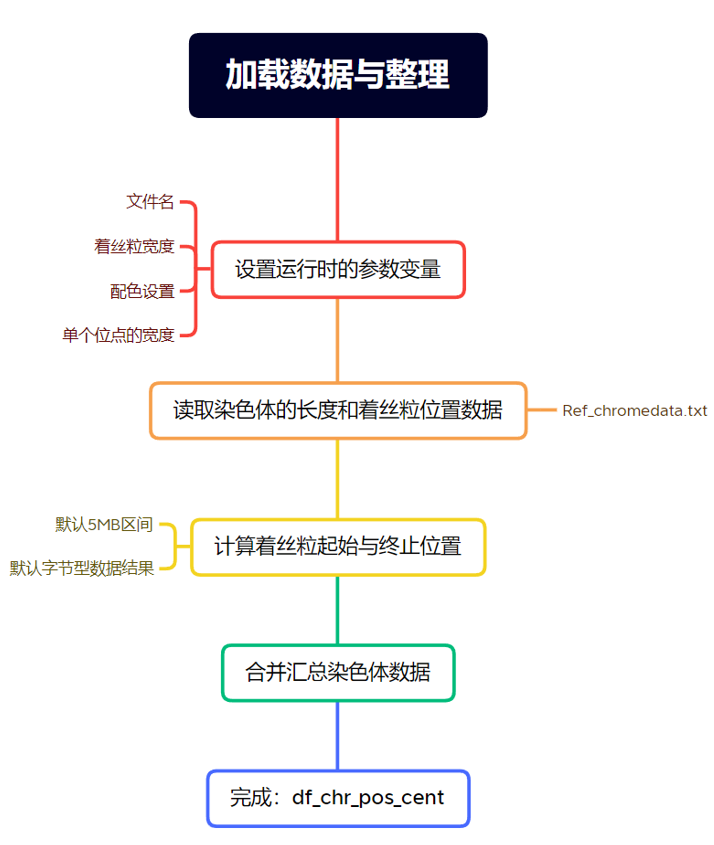
先从名为 "Ref_chromedata.txt" 的文件中读取数据,并将其存储在名为 df_chr_pos_cent 的数据框中,再选取某些列的内容,并将它们拼接成一个新的矩阵,自动计算每条染色体的起止位置和着丝粒的起止位置。
创建判定子代和亲本关系的函数
为了更方便的计算某个位点在亲本和子代中的存在情况,快速获得子代中突变信息的来源,创建一个亲缘关系判定函数。
# 函数:输入两个亲本和子代基因型,判断子代来源
determine_progeny <- function(p1,p2,prog,name_p1,name_p2){
if (p1 == p2){
if (p1 == prog & p2 == prog){
return("Both")
}else{
return("Unknown")
}
}else{
if (p1 == prog){
return(name_p1)
}else{
if (p2 == prog){
return(name_p2)
}else{
return("Unknown")
}
}
}
}
这个函数叫做 determine_progeny,它用于确定两个亲本(p1 和 p2)以及一个后代(prog)之间的关系。
参数说明:
-
p1 - 第一个亲本的标识。 -
p2 - 第二个亲本的标识。 -
prog - 后代的标识。 -
name_p1 - 第一个亲本的名字 -
name_p2 - 第二个亲本的名字
功能介绍:
首先,检查如果第一个亲本 p1 和第二个亲本 p2 相等,那么它们之间的关系有两种情况:
-
如果它们都等于后代 prog,那么返回 "Both",表示两个亲本都与后代相关。 -
另外一种情况,返回 "Unknown",表示无法确定它们之间的关系。
如果第一个亲本 p1 和第二个亲本 p2 不相等,那么它们之间的关系也有两种情况:
-
如果第一个亲本 p1 等于后代 prog,那么返回 name_p1,表示第一个亲本是后代的亲本。 -
如果第二个亲本 p2 等于后代 prog,那么返回 name_p2,表示第二个亲本是后代的亲本。
最后,如果以上条件都不满足,返回 "Unknown",表示无法确定它们之间的关系。 这个函数的目的是根据输入的亲本和后代信息,确定它们之间的关系,并返回一个相应的标注信息。
创建判定杂合子的函数
如果某些位点为杂合状态,需要对其进行识别标注,以下提供一种算法进行识别:
# 函数:判断杂合子
decide_hybrid <- function(genetype){
tem <- str_split(genetype,"")
if (length(tem[[1]]) != 2){
return("Error")
}else{
first <- tem[[1]][1]
sencend <- tem[[1]][2]
if (first == sencend){
return("equal")
}else{return("diff")}
}
}
这段代码定义了一个名为decide_hybrid的函数,其作用是接受一个字符串参数genetype,然后将该字符串拆分成两个字符,并检查这两个字符是否相等。
如果输入字符串的长度不等于2,函数将返回"Error",表示输入错误。
否则,如果两个字符相等,函数将返回"equal",表示相等。
如果两个字符不相等,函数将返回"diff",表示不相等。这个函数主要用于比较输入的基因型是否相等或不等。 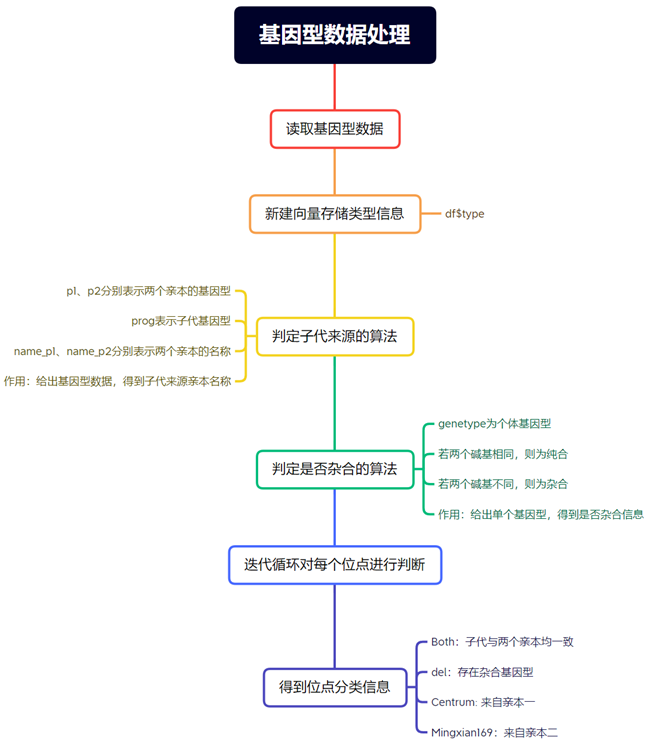
计算每个位点的变异类型
for (i in 1:nrow(df)){
if (df$chr[i] == "#N/A"){
df$type[i] <- "del"
next
}
if (df$pos[i] == "#N/A"){
df$type[i] <- "del"
next
}
if (decide_hybrid(df[i,3]) == "diff" |
decide_hybrid(df[i,4]) == "diff" |
decide_hybrid(df[i,5]) == "diff"){
df$type[i] <- "del"
next
}
df$type[i] <- determine_progeny(df[i,3],df[i,4],df[i,5],
colnames(df)[3],colnames(df)[4])
}
这段代码的主要作用是根据一系列条件来确定df 数据框中每一行的"type"列的值,并根据不同条件进行相应的处理。如果某些条件满足,它将类型设置为 "del",否则它会计算新的类型并将其存储在数据框中。
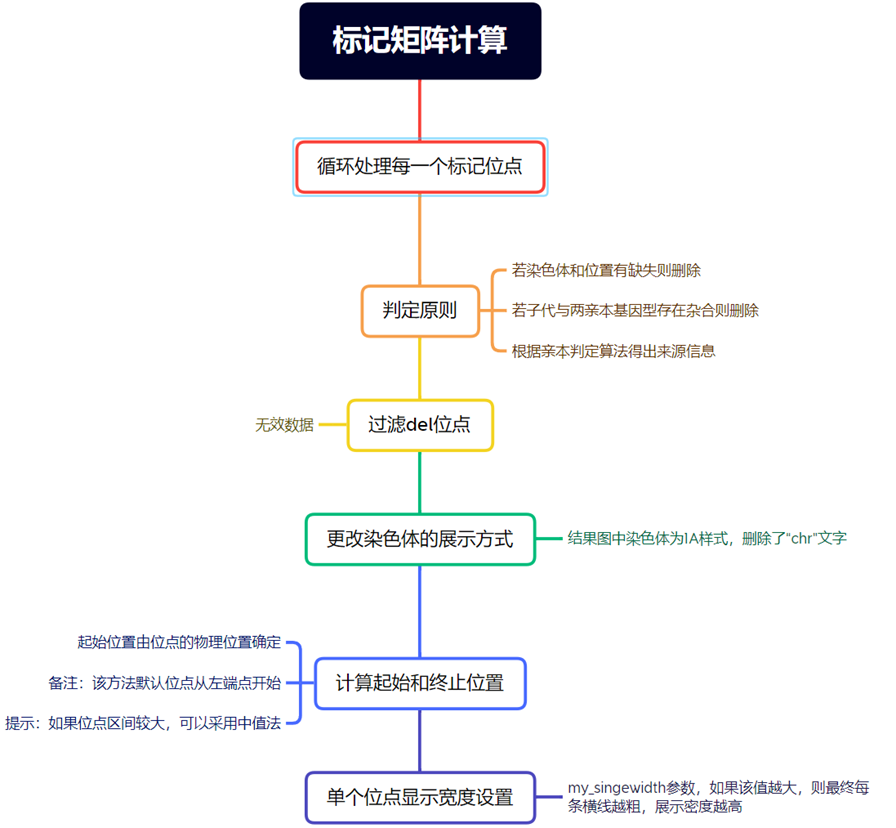
绘图数据初步整理
df_marker <- df %>% filter(type != "del")
df_marker$chr <- sub("chr","",df_marker$chr)
df_marker$Start <- df_marker$pos
df_marker$End <- as.numeric(df_marker$pos) + my_singewidth
df_marker$Value[which(df_marker$type == "Both")] <- 4
df_marker$Value[which(df_marker$type == colnames(df)[3])] <- 3
df_marker$Value[which(df_marker$type == colnames(df)[4])] <- 2
df_marker$Value[which(df_marker$type == "Unknown")] <- 1
首先从数据框df中筛选出type列不等于"del"的行,然后对筛选后的数据框df_marker进行一系列操作:
移除df_marker$chr列中的"chr"前缀,将结果存储回df_marker$chr中。 将df_marker$pos列的值赋给新的列df_marker$Start,以便将位置信息重命名。 根据my_singewidth的值计算新的列df_marker$End,该列的值等于df_marker$pos加上my_singewidth的数值。
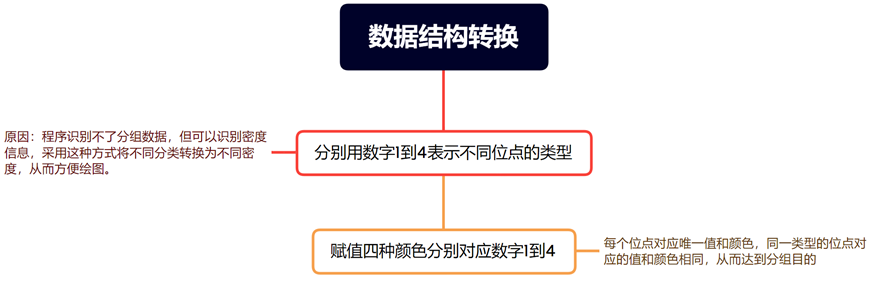
根据条件对df_marker$Value列进行更新,将根据type列的不同值分配不同的数值,以下为对应规则:
如果type等于"Both",则将df_marker$Value设置为4。
如果type等于数据框中的第三列的列名,则将df_marker$Value设置为3。
如果type等于数据框中的第四列的列名,则将df_marker$Value设置为2。
如果type等于"Unknown",则将df_marker$Value设置为1。
主要目的是根据条件对df数据框进行筛选和转换操作,创建一个数据框df_marker,其中包括重命名列、计算新列以及根据不同的type值赋予相应的数值。
转换数据格式
df_marker_plot <- cbind(df_marker[,c(1,7,8,9)])
df_marker_plot <- df_marker_plot %>%
mutate_at(vars(Start,End,Value),as.integer)
df_marker_plot <- df_marker_plot[which(df_marker_plot$chr !="Un"),]
colnames(df_marker_plot) <- c("Chr","Start","End","Value")
这段代码的功能是对名为df_marker的数据框进行处理,首先选择其中的特定列(第1、7、8、9列),然后将这些列的数据类型转换为整数类型,接着从中删除chr列值不等于"Un"的行,最后重命名这些列为"Chr"、"Start"、"End"和"Value",用于后续的数据可视化或分析。
绘制染色体全景图
# 绘制全景图
ideogram(karyotype = df_chr_pos_cent,
overlaid = df_marker_plot,
colorset1 = my_color)
convertSVG("chromosome.svg", device = "pdf")
最后,使用如上代码即可生成最终的结果图片。该R包非常巧妙的使用SVG语法构建图形,并且提供了svg转pdf、png等多种方式。
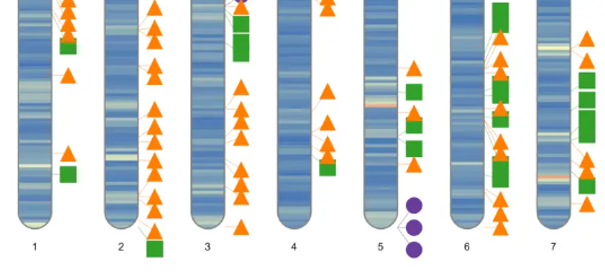
提示:分类型离散数据通过数值映射转换为连续型,便可以通过函数进行绘图计算,如果有多个维度的数据需要展示,可以添加其他类型的标注。
多组学应用:
-
差异表达基因的分布(RNA-seq) -
开放染色质的分布(ATAC-seq) -
CTCF结合位点(ChIP-seq) -
突变位点在染色体上的分布(WGS) -
DNA甲基化的分布(WGBS) -
外显子在染色体上的分布(WES)
遗传学应用:
-
变异位点全景图绘制 -
标记物理位置展示图绘制 -
遗传多样性来源展示 -
基因连锁定位结果展示
>>> 参考资料
https://github.com/cran/RIdeogram
http://doi.org/10.7717/peerj-cs.251
https://www.jianshu.com/p/07ae1fe18071
>>> Tips:本文所有示例数据均随机生成,不具有任何意义
本文由 mdnice 多平台发布
相关文章:
R语言绘制染色体变异位置分布图,RIdeogram包
变异位点染色体分布图 今天分享的内容是通过RIdeogram包绘制染色体位点分布图,并介绍一种展示差异位点的方法。 在遗传学研究中,通过测序等方式获得了基因组上某些位置的基因型信息。 如下表,第一列是变异位点的ID,第二列是染色体…...
每天10个小知识点)
Vue知识系列(7)每天10个小知识点
目录 系列文章目录Vue知识系列(1)每天10个小知识点Vue知识系列(2)每天10个小知识点Vue知识系列(3)每天10个小知识点Vue知识系列(4)每天10个小知识点Vue知识系列(5&#x…...
5分钟就能实现的API监控,有什么理由不做呢?
API深度影响着你的应用 今天的数字应用世界其实是一个以API为中心的世界,我们只是没有意识到这些API的重要性。比如在电子商务交易、社交媒体等对交互高度依赖的领域,可以说API决定了应用的质量一点也不为过。 以京东为例,用户的每一次操作背…...
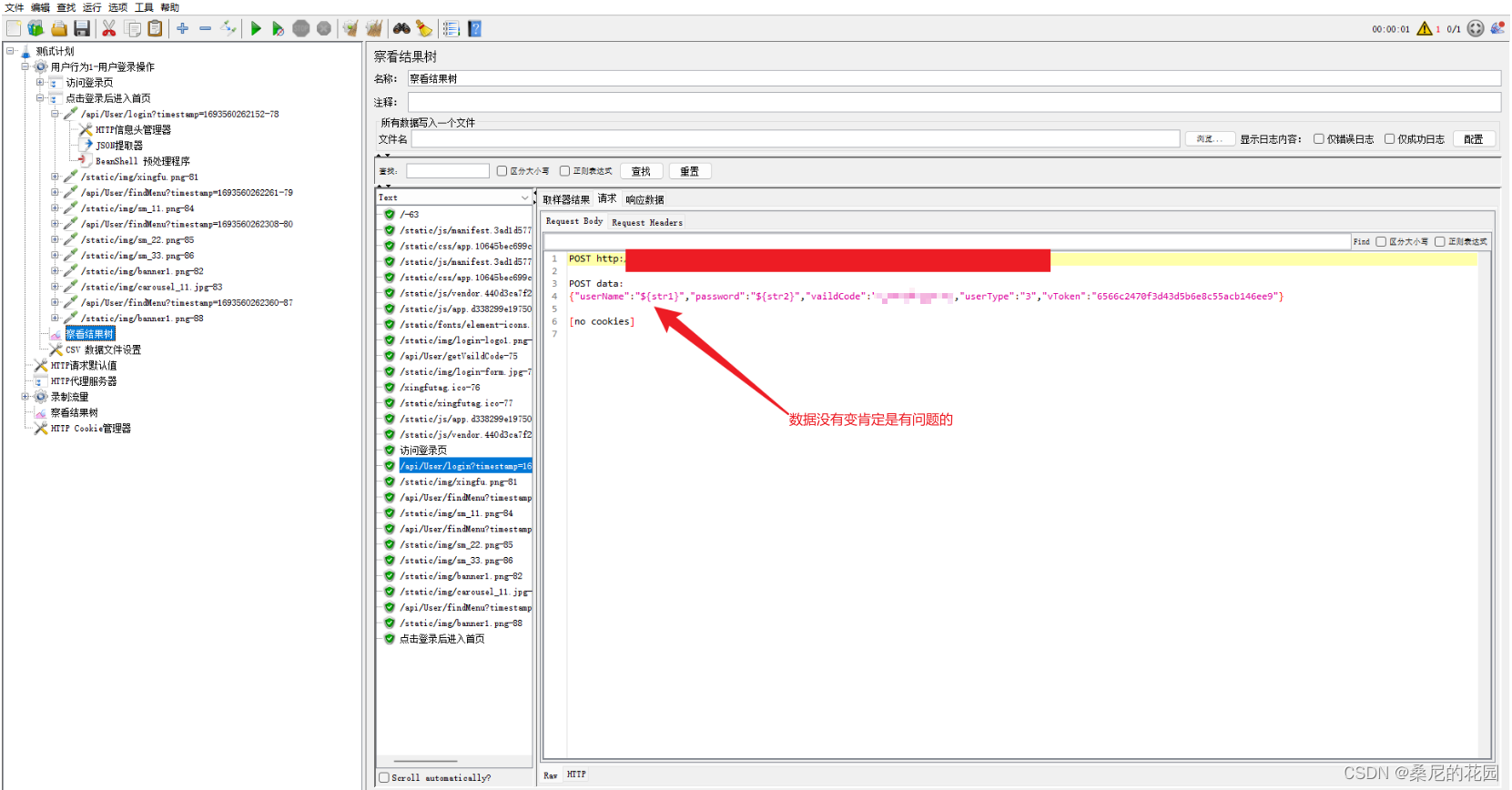
Jmeter引入外部jar包以满足加密数据的Post请求
目录 一、把项目打成jar包 1、创建一个Maven项目,并保证可以正常运行。 2、把工具类放置项目中,确保无报错且能够正常使用。 3、打包 4、验证 jar包是否有效 5、你想打多个工具类的包 二、在jmeter中使用 1、把jar包放到jmeter仓库下,…...

了解冒泡排序
package com.mypackage.array;import java.util.Arrays;public class Demo07 {public static void main(String[] args) {int[] a {3,2,6,7,4,5,6,34,56,7};int[] sort1 sort1(a); //调用我们自己写的排序方法后,返回一个排序后的数组System.out.println(Array…...
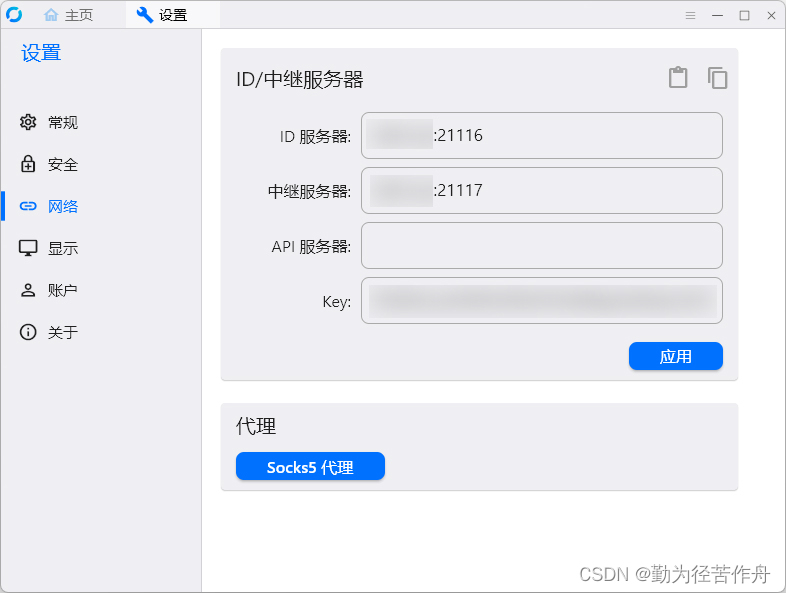
群辉 Synology NAS Docker 安装 RustDesk-server 自建服务器只要一个容器
from https://blog.zhjh.top/archives/M8nBI5tjcxQe31DhiXqxy 简介 之前按照网上的教程,rustdesk-server 需要安装两个容器,最近想升级下版本,发现有一个新镜像 rustdesk-server-s6 可以只安装一个容器。 The S6-overlay acts as a supervi…...
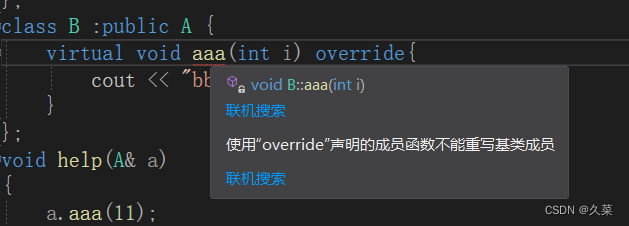
为什么要有override
多态一定会成功吗 因为逻辑是用户编写的,那么肯定会有遗漏的地方,那就要规则来限制。就比如多态,都知道条件之一是子类重写了父类的虚函数,但是如果子类没有严格遵守这个规则,就无法达到目的。就比如这个代码…...

Linux界的老古董
Slackware 是由 Patrick Volkerding 制作的 Linux 发行版,从 1993 年发布至今也一直在 Patrick 带领下进行维护。7 月 17 日,Slackware 才刚刚过完它 24 岁的生日,看似年纪轻轻的它,已然是 Linux 最古老的发行版。 Slackware 的发…...

安卓逆向 - Xposed入门教程
一、引言 Xposed框架,是Android中Hook技术的一个著名的框架,拥有非常丰富的模块,给我们分析app提供了极大的便利,Xposed框架是开源的。最高支持到Android 8(重要) github地址:GitHub - rovo89…...

【嵌入式】2024届校招岗位汇总
公司岗位博世嵌入式自动化测试工程师博世嵌入式开发(软件刷写及启动)工程师博世Linux/C软件工程师博世自动驾驶软件开发工程师博世嵌入式软件工程师(BSP)博世嵌入式电子工程师 (BMS&电源)博世物联网嵌入式开发工程师 …...
Docker搭建ELK日志采集服务及Kibana可视化图表展示
架构 ES docker network create elkmkdir -p /opt/ELK/es/datachmod 777 /opt/ELK/esdocker run -d --name elasticsearch --net elk -p 9200:9200 -p 9300:9300 -e "discovery.typesingle-node" -v /opt/ELK/es/plugins:/usr/share/elasticsearch/plugins -v /opt/…...

SpringBoot结合MyBatis实现多数据源配置
SpringBoot结合MyBatis实现多数据源配置 一、前提条件 1.1、环境准备 SpringBoot框架实现多数据源操作,首先需要搭建Mybatis的运行环境。 由于是多数据源,也就是要有多个数据库,所以,我们创建两个测试数据库,分别是…...

单个vue echarts页面
<template> <div ref"history" class"echarts"></div> </template> <script> export default{ data () { return {}; }, methods: { history(){ let myChart this.$echarts.init(this.$refs.history); // 绘制图表 myCha…...
【web开发】6、Django(1)
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档 文章目录 一、Django是什么?二、使用步骤1.安装Django2.创建项目3.创建app4.快速上手5.模板继承 数据库操作1.安装第三方模块2.自己创建数据库3.DJango链接数据库…...
第29节-PhotoShop基础课程-滤镜库
文章目录 前言1.滤镜库2.Camera Raw滤镜 (用来对图片进行预处理,最全面的一个)3.神经滤镜(2022插件 需要先下载)4.液化(胖-> 瘦 矮->高)5.其它滤镜1.自适应广角2.镜头矫正 把图片放正3.消…...
)
空间(蓝桥杯)
空间 本题为填空题,只需要算出结果后,在代码中使用输出语句将所填结果输出即可。 小蓝准备用 256MB 的内存空间开一个数组,数组的每个元素都是 32 位 二进制整数,如果不考虑程序占用的空间和维护内存需要的辅助空间,…...
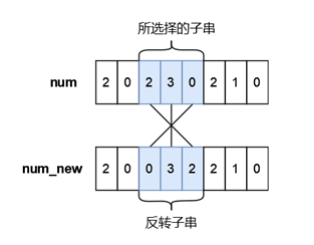
蓝桥杯2023年第十四届省赛真题-更小的数--题解
目录 蓝桥杯2023年第十四届省赛真题-更小的数 题目描述 输入格式 输出格式 样例输入 样例输出 提示 【思路解析】 【代码实现】 蓝桥杯2023年第十四届省赛真题-更小的数 时间限制: 3s 内存限制: 320MB 提交: 895 解决: 303 题目描述 小蓝有一个长度均为 n 且仅由数字…...

SpringBoot详解
文章目录 SpringBoot的特点Spring,SpringBoot的区别SpringBoot常用注解标签SpringBoot概述SpringBoot简单Demo搭建读取配置文件的内容 SpringBoot自动配置Condition自定义beanSpringBoot常用注解原理EnableAutoConfiguration SpringBoot监听机制SpringBoot启动流程分…...

typescript 类型断言
typescript 类型断言 TypeScript 是一种在 JavaScript 基础上开发的强类型语言,它为开发者提供了类型安全性和其他有用的特性。类型断言是 TypeScript 中的一种特性,允许开发者在编译时确定变量或表达式的类型。类型断言有多种使用场景,包括…...

如何确定自己是否适合做程序员?
如果你不确定你是否注定要成为一名程序员,这里有六个迹象可能表明你不适合。 1. 你缺乏实验创造力 尽管编程的基础是逻辑,但它在很大程度上是一种创造性的艺术。新程序就像一张空白的画布。画笔和调色板是语言、框架和库。您需要对自己的创作和创造力有…...

从栅格到矢量:基于ArcScan的河道中心线智能提取与精度优化实践
1. 从栅格到矢量的技术背景 河道中心线提取是水文分析中的基础性工作。传统人工勾绘方式效率低下,一条10公里长的河道可能需要耗费专业人员半天时间。而基于ArcScan的自动化提取方法,能将这个时间缩短到10分钟以内,同时保证亚米级精度。 我在…...
)
YouTube长视频摘要失效真相(Gemini模型行为深度逆向分析)
更多请点击: https://intelliparadigm.com 第一章:YouTube长视频摘要失效真相(Gemini模型行为深度逆向分析) 近期大量开发者反馈 YouTube 长视频(>30 分钟)通过 Gemini API 生成的摘要内容空洞、重复或…...

百度网盘Mac破解终极方案:解锁SVIP高速下载体验
百度网盘Mac破解终极方案:解锁SVIP高速下载体验 【免费下载链接】BaiduNetdiskPlugin-macOS For macOS.百度网盘 破解SVIP、下载速度限制~ 项目地址: https://gitcode.com/gh_mirrors/ba/BaiduNetdiskPlugin-macOS 作为Mac用户,你是否曾因百度网盘…...

给视觉开发新手的保姆级教程:在Ubuntu上从下载源码到成功运行Demo,搞定OpenCV 3环境搭建
给视觉开发新手的保姆级教程:在Ubuntu上从下载源码到成功运行Demo,搞定OpenCV 3环境搭建 第一次在Ubuntu上搭建OpenCV开发环境,对很多视觉开发新手来说可能是个令人望而生畏的任务。命令行操作、编译工具链、环境配置……这些术语听起来就让人…...

基于MCP协议与本地全文检索的电子元件文档AI查询系统
1. 项目概述:为LLM构建一个本地化的电子元件文档搜索引擎如果你是一名嵌入式工程师、硬件开发者,或者像我一样,经常需要和德州仪器(TI)、意法半导体(ST)、亚德诺(ADI)这些…...

避开这些坑:ADSP-SC589开发中JTAG连接、驱动安装与调试的常见问题解决
ADSP-SC589开发实战:JTAG连接与调试避坑指南 当ADSP-SC589开发板与AD-HP530ICE仿真器首次相遇时,许多开发者会陷入连接失败的困境。不同于普通MCU开发,SHARC系列DSP的JTAG调试存在诸多技术细节,稍有不慎就会导致数小时的无效排查。…...
)
卸载软件后右键菜单残留?用PowerShell精准清理注册表(附一键备份脚本)
彻底告别右键菜单残留:PowerShell注册表清理实战指南 刚卸载完某款压缩软件,却发现右键菜单里依然顽固地留着它的选项——这种经历恐怕不少Windows用户都遇到过。上周帮同事处理电脑时,就遇到了一个典型案例:卸载"可牛压缩&q…...

9.实战案例拆解
好的,我们开始。先别急着看那些“月入十万”的爽文,我这边先给你看一段我昨晚在调试一个树莓派Pico W的I2C总线时,在终端里敲出来的报错信息: [ERROR] I2C timeout: SDA line held low by device at 0x3C这条错误让我折腾了半小时。最后发现是传感器模块的电源纹波太大,导…...

Dism++终极指南:5步彻底解决Windows系统卡顿和臃肿问题
Dism终极指南:5步彻底解决Windows系统卡顿和臃肿问题 【免费下载链接】Dism-Multi-language Dism Multi-language Support & BUG Report 项目地址: https://gitcode.com/gh_mirrors/di/Dism-Multi-language 你是否曾为Windows系统越来越慢而烦恼…...

3D Tiles-Tools实战指南:如何高效处理大规模地理空间3D数据转换?
3D Tiles-Tools实战指南:如何高效处理大规模地理空间3D数据转换? 【免费下载链接】3d-tiles-tools 项目地址: https://gitcode.com/gh_mirrors/3d/3d-tiles-tools 在数字孪生、智慧城市和地理信息系统领域,大规模3D地理空间数据的高效…...