【R包】pathlinkR转录组数据分析和可视化利器

介绍
通常情况下,基因表达研究如微阵列和RNA-Seq会产生数百到数千个差异表达基因(deg)。理解如此庞大的数据集的生物学意义变得非常困难,尤其是在分析多个条件和比较的情况下。该软件包利用途径富集和蛋白-蛋白相互作用网络,促进了差异基因表达结果的可视化和下游分析,帮助研究人员从基因表达研究中发现潜在的生物学和病理生理学。
我们在这个包中包含了一个基因表达结果的示例数据集,作为对象“exampleDESeqResults”。这是一个由2个数据帧组成的列表,使用包中的‘ results() ’函数生成BiocStyle::Biocpkg(“DESeq2”)’ (Love et al. 2014)。数据来自一项RNA-Seq研究,该研究调查了入院时(T1)的COVID-19和非COVID-19败血症患者与大约1周后(T2)在ICU的情况,并随时间索引(即T2 vs T1) (an et al. 2023)。
安装
安装该R包
# We'll also be using some functions from dplyr
# BiocManager::install("pathlinkR", version="devel")
library(dplyr)
library(pathlinkR)
基因表达火山图
在基因表达研究中,通常首先进行的可视化分析之一是识别差异表达基因(DEGs)的数量。这些基因通常是通过设定特定的倍数变化和统计显著性阈值来定义的。调整后的p值小于0.05和绝对倍数变化大于1.5被用作默认阈值,尽管可以指定任何值。pathlinkR 包含了一个名为 eruption() 的函数,用于创建火山图。
data("exampleDESeqResults")eruption(rnaseqResult=exampleDESeqResults[[1]],title=names(exampleDESeqResults[1])
)

对于定制火山图,有多种选项可供选择,包括:
- 调整阈值
- 切换显著基因和非显著基因的颜色
- 改变标记基因的数量
- 调整x轴和y轴的范围
- 绘制对数2(log2)或非对数2的倍数变化(以更好地感知变化幅度)
- 突出显示特定感兴趣的基因集
data("sigoraDatabase")
interferonGenes <- sigoraDatabase %>% filter(pathwayName == "Interferon Signaling") %>% pull(ensemblGeneId)eruption(rnaseqResult=exampleDESeqResults[[1]],title=names(exampleDESeqResults[1]),highlightGenes=interferonGenes,highlightName="Interferon genes (red)",label="highlight",nonlog2=TRUE,n=10
)

可视化比较中的倍数变化
除了创建火山图外,我们还可以通过热图来可视化特定通路(例如通过pathwayEnrichment()识别为显著的通路)中涉及的差异表达基因(DEGs)。plotFoldChange()函数通过接收DESeq2::results()数据框的输入列表(与pathwayEnrichment()类似),并为组成基因的倍数变化创建热图来实现这一点。
plotFoldChange(inputList=exampleDESeqResults,pathName="Interferon alpha/beta signaling"
)

为了定制化,提供了多种选项,包括:
- 缩短每次比较的名称
- 提供一个自定义的基因列表进行可视化,并添加一个信息性的标题
- 在热图单元格中使用对数2倍数变化
- 交换默认的行和列,并在行(比较)之间添加分隔
exampleDESeqResultsRenamed <- setNames(exampleDESeqResults,c("Pos", "Neg")
)plotFoldChange(exampleDESeqResultsRenamed,manualTitle="Signature genes",genesToPlot=c("CD4", "CD8A","CD8B", "CD28", "ZAP70"),geneFormat="hgnc",colSplit=c("Pos", "Neg"),log2FoldChange=TRUE,colAngle=45,clusterRows=FALSE,clusterColumns=TRUE,invert=TRUE
)

构建和可视化蛋白质-蛋白质相互作用网络
pathlinkR 包含用于构建和可视化蛋白质-蛋白质相互作用(PPI)网络的工具。在这里,我们利用从 InnateDB 收集的 PPI 数据,生成在基因表达分析中识别的差异表达基因(DEGs)之间的相互作用列表。这些相互作用随后可以在 R 中用于构建 PPI 网络,同时提供了多种选项来控制网络的类型,例如支持一级、最小或零级网络。实现这一目标的两个主要函数是 ppiBuildNetwork() 和 ppiPlotNetwork()。
让我们继续查看 COVID 阳性患者随时间变化的差异表达基因(DEGs),使用显著的 DEGs 来构建一个 PPI 网络。由于我们输入的数据框包含了所有测量的基因(而不仅仅是显著的基因),我们将使用 filterInput=TRUE 选项,以确保网络仅由通过标准阈值(如上所述)的基因构建。由于我们正在可视化 DEGs 的网络,让我们通过指定 fillType="foldChange" 来给节点上色,以指示它们失调的方向(即上调或下调)。
exNetwork <- ppiBuildNetwork(rnaseqResult=exampleDESeqResults[[1]],filterInput=TRUE,order="zero"
)ppiPlotNetwork(network=exNetwork,title=names(exampleDESeqResults)[1],fillColumn=LogFoldChange,fillType="foldChange",label=TRUE,labelColumn=hgncSymbol,legend=TRUE
)

带有蓝色标签的节点(例如STAT1、FBXO6、CDH1等)是网络中的枢纽节点,即那些具有高介数中心性得分的基因。用于确定枢纽节点的统计量可以在ppiBuildNetwork()函数中通过“hubMeasure”选项进行设置。
exNetworkInterferon <- mutate(exNetwork,isInterferon = if_else(name %in% interferonGenes, "y", "n")
)ppiPlotNetwork(network=exNetworkInterferon,title=names(exampleDESeqResults)[1],fillColumn=isInterferon,fillType="categorical",catFillColours=c("y"="red", "n"="grey"),label=TRUE,labelColumn=hgncSymbol,legend=TRUE,legendTitle="Interferon\ngenes"
)

富集网络和提取子网络
pathlinkR 包含两个用于进一步分析蛋白质-蛋白质相互作用(PPI)网络的函数。首先,ppiEnrichNetwork() 函数会使用网络的节点表来测试富集的 Reactome 通路或 Hallmark 基因集。
exNetworkPathways <- ppiEnrichNetwork(network=exNetwork,analysis="hallmark",filterResults="default",geneUniverse = rownames(exampleDESeqResults[[1]])
)exNetworkPathways

其次,ppiExtractSubnetwork() 函数可以从起始网络中提取一个最小连通的子网络,使用富集通路中的基因作为提取的“起始”节点。例如,下面我们将使用上述 Hallmark 富集的结果,从“干扰素γ反应”项中提取一个基因子网络,然后绘制这个缩减后的网络,并突出显示通路中的基因。
exSubnetwork <- ppiExtractSubnetwork(network=exNetwork,pathwayEnrichmentResult=exNetworkPathways,pathwayToExtract="INTERFERON GAMMA RESPONSE"
)ppiPlotNetwork(network=exSubnetwork,fillType="oneSided",fillColumn=degree,label=TRUE,labelColumn=hgncSymbol,legendTitle="Degree"
)

或者,您也可以使用ppiExtractSubnetwork()函数中的genesToExtract参数,提供您自己的基因集(一个包含Ensembl ID的字符向量)来提取为子网络。
进行通路富集分析
通路富集的核心概念是过度表达:也就是说,我们差异表达基因(DEG)列表中属于特定通路的基因数量是否比随机情况下预期的更多?为了计算这一点,最简单的方法是比较某个通路中的 DEGs 与所有 DEGs 的比例,以及数据库中所有通路中的基因与该通路中所有基因的比例。pathlinkR 主要使用 Reactome 数据库(Fabregat 等,2017)来实现这一目的。
过度表达分析可能出现的一个问题是,假设每个通路中的每个基因在属于该通路方面具有“相等”的价值。实际上,一种蛋白质可以具有多种(有时是非常不同的)功能,并且属于多个通路,例如蛋白激酶。还有一些通路与细胞机制有相当大的重叠,如 TLR 通路。这可能导致多个相似通路或甚至不相关的“假阳性”富集,使得解析结果变得非常困难。
一种解决方案是使用独特的基因对,正如 r BiocStyle::CRANpkg("sigora") 包的创建者所描述的(Foroushani 等,2013)。这种方法减少了由于多面性基因导致的相似和不相关通路的数量,更多地关注可能与基础生物学相关的通路。这种方法是 pathlinkR 中 pathwayEnrichment() 函数的默认方法。
pathwayEnrichment() 函数的输入是一个数据框列表(每个来自 DESeq2::results()),默认情况下会将基因分为上调和下调两类,然后分别对每组进行通路富集分析。列表中数据框的名称应表明在 DESeq2 结果中进行的比较,因为它将用于识别结果。对于 analysis="sigora",我们还需要提供一个基因对签名库(gpsRepo),其中包含要测试的通路和基因对。将此参数保留为“默认”将使用 sigora 中的 reaH GPS 库,包含人类 Reactome 通路。或者,也可以提供自己的 GPS 库;有关如何制作的详细信息,请参阅 ??sigora::makeGPS()。
## Note the structure of `exampleDESeqResults`: a named list of results from
## DESeq2
exampleDESeqResultsenrichedResultsSigora <- pathwayEnrichment(inputList=exampleDESeqResults,analysis="sigora",filterInput=TRUE,gpsRepo="default"
)head(enrichedResultsSigora)

对于那些仍然倾向于传统的过度表达分析的人,我们提供了通过设置analysis="reactome"来使用r BiocStyle::Biocpkg("ReactomePA")(Yu 等,2016)进行分析的选项。在使用这种方法时,我们建议提供一个基因全集,作为富集测试的背景;这里我们将使用 DESeq2 测试显著性的所有基因(即计数矩阵中的所有基因),在运行测试之前将它们转换为 Entrez 基因 ID。详情请参阅我们的 Github 页面 上的完整手册。
除了在设置analysis为“sigora”或“reactome”时使用的 Reactome 数据库外,我们还提供了使用 Molecular Signatures Database (MSigDb) 中的 Hallmark 基因集 进行过度表达分析。这些是 50 个基因集,代表“具有协调表达的特定、明确定义的生物学状态或过程”(Liberzon 等,2015)。与更细致的 Reactome 通路相比,这个数据库提供了关于关键生物学过程的更高级别的总结
enrichedResultsHm <- pathwayEnrichment(inputList=exampleDESeqResults,analysis="hallmark",filterInput=TRUE,split=TRUE
)head(enrichedResultsHm)

最后,用户在进行富集分析时也可以使用来自 KEGG 的数据。通过设置analysis="kegg",可以使用传统的过度表达分析;或者通过指定analysis="sigora"和gpsRepo="kegH",可以使用基于基因对的方法。
绘制通路富集分析结果
现在我们已经得到了来自多个比较的大量通路富集分析结果,是时候将它们可视化了。plotPathways()函数通过将Reactome通路(或Hallmark基因集)分组到父组下,并指示每个通路在每个比较中是上调还是下调,从而帮助我们轻松识别哪些通路在不同的差异表达基因(DEG)列表中是共享的或独特的。由于通常会有许多通路,您可以将绘图分成多列(最多3列),并截断通路名称以使结果更容易展示。
有时,一个通路可能在同一个DEG列表中的上调和下调基因中都富集(这种情况通常发生在较大的通路中)。这种现象会用一个白色星号表示,其中显示更显著(调整后的p值更低)的方向。您还可以更改比较的角度/标签,或者在标签下方添加每个比较中的DEG数量。最后,您可以指定要包含在可视化的通路或顶级通路组。
pathwayPlots(pathwayEnrichmentResults=enrichedResultsSigora, columns=2
)

这些绘图也可以进行多种调整:
- 基于顶级通路分组,仅显示与免疫相关的通路
- 改变用于调整后p值的颜色缩放
- 压缩比较名称以便更好地适应绘图,并使其水平显示
- 在比较名称下方添加用于富集的DEGs数量
- 增加通路名称的截断截止值,以便显示更多单词
从这些结果中,你可以看到,尽管许多免疫系统通路在COVID-19和非COVID-19脓毒症患者中随时间朝同一方向变化,但有一些独特的通路脱颖而出,大多与干扰素信号传导有关(“干扰素信号传导”、“干扰素γ信号传导”、“干扰素α/β信号传导”、“ISG15抗病毒机制”)。这可能反映了COVID-19患者早期抗病毒反应的增强,这种反应随时间减弱,而非COVID-19脓毒症患者则没有变化。
pathwayPlots(pathwayEnrichmentResults=enrichedResultsSigora,specificTopPathways="Immune System",colourValues=c("#440154", "#FDE725"),newGroupNames=c("COVID\nPositive", "COVID\nNegative"),showNumGenes=TRUE,xAngle="horizontal",nameWidth=50
)

从富集通路生成网络
pathlinkR 包含将基于 Reactome 的方法(“sigora”或“reactomepa”)的通路富集结果转换为网络的函数,通过计算分配给每个通路的基因的重叠来确定它们之间的相似性。在这些网络中,每个通路是一个节点,它们之间的连接或边是通过距离度量来确定的。可以设置一个阈值,当两个通路之间的相似性达到最小值时,它们被认为是相连的,并且会在它们的节点之间绘制一条边。
我们提供了一个预先计算好的 Reactome 通路距离矩阵,该矩阵是使用 Jaccard 距离生成的,但也支持使用多种距离度量。一旦创建了这个通路相互作用的“基础”,就可以使用 createPathnet() 函数构建通路网络:
data("sigoraDatabase")pathwayDistancesJaccard <- getPathwayDistances(pathwayData = sigoraDatabase)startingPathways <- pathnetFoundation(mat=pathwayDistancesJaccard,maxDistance=0.8
)# Get the enriched pathways from the "COVID Pos Over Time" comparison
exPathwayNetworkInput <- enrichedResultsSigora %>% filter(comparison == "COVID Pos Over Time")myPathwayNetwork <- pathnetCreate(pathwayEnrichmentResult=exPathwayNetworkInput,foundation=startingPathways
)
另一种可视化方法
pathnetGGraph(myPathwayNetwork,labelProp=0.1,nodeLabelSize=3,nodeLabelOverlaps=8,segColour="red",themeBaseSize = 12
)

填充的节点(通路)是富集的通路(即由pathwayEnrichment()输出的通路)。节点的大小与统计显著性相关,而边的厚度与两个相连通路的相似性有关。
pathnetVisNetwork(myPathwayNetwork)

仅使用差异表达基因(DEGs)生成通路网络
另一种选择是在生成通路网络信息时仅使用差异表达基因(DEGs),具体方法如下:
candidateData <- sigoraExamples %>%filter(comparison == "COVID Pos Over Time") %>% select(pathwayId, genes) %>%tidyr::separate_rows(genes, sep=";") %>%left_join(sigoraDatabase,by=c("pathwayId", "genes" = "hgncSymbol"),multiple="all") %>%relocate(pathwayId, ensemblGeneId, "hgncSymbol"=genes, pathwayName) %>%distinct()## Now that we have a smaller table in the same format as sigoraDatabase, we
## can construct our own matrix of pathway distances
candidateDistData <- getPathwayDistances(pathwayData=candidateData,distMethod="jaccard"
)candidateStartingPathways <- pathnetFoundation(mat=candidateDistData,maxDistance=0.9
)candidatesAsNetwork <- pathnetCreate(pathwayEnrichmentResult=filter(sigoraExamples, comparison == "COVID Pos Over Time"),foundation=candidateStartingPathways,trim=FALSE
)pathnetVisNetwork(candidatesAsNetwork, nodeLabelSize=30)

系统信息
R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.5 LTSMatrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0locale:[1] LC_CTYPE=en_CA.UTF-8 LC_NUMERIC=C [3] LC_TIME=en_CA.UTF-8 LC_COLLATE=en_CA.UTF-8 [5] LC_MONETARY=en_CA.UTF-8 LC_MESSAGES=en_CA.UTF-8 [7] LC_PAPER=en_CA.UTF-8 LC_NAME=C [9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_CA.UTF-8 LC_IDENTIFICATION=C time zone: America/Vancouver
tzcode source: system (glibc)attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base other attached packages:[1] DESeq2_1.46.0 SummarizedExperiment_1.36.0[3] Biobase_2.66.0 MatrixGenerics_1.18.0 [5] matrixStats_1.4.1 GenomicRanges_1.58.0 [7] GenomeInfoDb_1.42.0 IRanges_2.40.0 [9] S4Vectors_0.44.0 BiocGenerics_0.52.0
[11] pathlinkR_1.2.0 dplyr_1.1.4
[13] BiocStyle_2.34.0 loaded via a namespace (and not attached):[1] RColorBrewer_1.1-3 rstudioapi_0.17.1 jsonlite_1.8.9 [4] shape_1.4.6.1 magrittr_2.0.3 ggtangle_0.0.4 [7] magick_2.8.5 farver_2.1.2 rmarkdown_2.28 [10] GlobalOptions_0.1.2 fs_1.6.5 zlibbioc_1.52.0 [13] vctrs_0.6.5 memoise_2.0.1 ggtree_3.14.0 [16] rstatix_0.7.2 tinytex_0.53 htmltools_0.5.8.1 [19] S4Arrays_1.6.0 broom_1.0.7 Formula_1.2-5 [22] gridGraphics_0.5-1 SparseArray_1.6.0 sass_0.4.9 [25] bslib_0.8.0 htmlwidgets_1.6.4 plyr_1.8.9 [28] cachem_1.1.0 igraph_2.1.1 lifecycle_1.0.4 [31] iterators_1.0.14 pkgconfig_2.0.3 gson_0.1.0 [34] Matrix_1.7-1 R6_2.5.1 fastmap_1.2.0 [37] GenomeInfoDbData_1.2.13 clue_0.3-65 aplot_0.2.3 [40] enrichplot_1.26.1 digest_0.6.37 colorspace_2.1-1 [43] patchwork_1.3.0 AnnotationDbi_1.68.0 RSQLite_2.3.7 [46] ggpubr_0.6.0 vegan_2.6-8 labeling_0.4.3 [49] fansi_1.0.6 httr_1.4.7 polyclip_1.10-7 [52] abind_1.4-8 mgcv_1.9-1 compiler_4.4.1 [55] bit64_4.5.2 withr_3.0.2 doParallel_1.0.17 [58] backports_1.5.0 BiocParallel_1.40.0 carData_3.0-5 [61] viridis_0.6.5 DBI_1.2.3 highr_0.11 [64] ggforce_0.4.2 R.utils_2.12.3 ggsignif_0.6.4 [67] MASS_7.3-61 DelayedArray_0.32.0 rjson_0.2.23 [70] permute_0.9-7 tools_4.4.1 ape_5.8 [73] R.oo_1.26.0 glue_1.8.0 nlme_3.1-166 [76] GOSemSim_2.32.0 grid_4.4.1 reshape2_1.4.4 [79] cluster_2.1.6 fgsea_1.32.0 generics_0.1.3 [82] gtable_0.3.6 R.methodsS3_1.8.2 tidyr_1.3.1 [85] data.table_1.16.2 car_3.1-3 tidygraph_1.3.1 [88] utf8_1.2.4 XVector_0.46.0 ggrepel_0.9.6 [91] foreach_1.5.2 pillar_1.9.0 stringr_1.5.1 [94] yulab.utils_0.1.7 circlize_0.4.16 splines_4.4.1 [97] tweenr_2.0.3 treeio_1.30.0 lattice_0.22-6
[100] bit_4.5.0 tidyselect_1.2.1 GO.db_3.20.0
[103] ComplexHeatmap_2.22.0 locfit_1.5-9.10 Biostrings_2.74.0
[106] knitr_1.48 gridExtra_2.3 bookdown_0.41
[109] xfun_0.49 graphlayouts_1.2.0 visNetwork_2.1.2
[112] stringi_1.8.4 UCSC.utils_1.2.0 lazyeval_0.2.2
[115] ggfun_0.1.7 yaml_2.3.10 evaluate_1.0.1
[118] codetools_0.2-20 ggraph_2.2.1 tibble_3.2.1
[121] qvalue_2.38.0 BiocManager_1.30.25 ggplotify_0.1.2
[124] cli_3.6.3 munsell_0.5.1 jquerylib_0.1.4
[127] Rcpp_1.0.13 png_0.1-8 parallel_4.4.1
[130] ggplot2_3.5.1 blob_1.2.4 clusterProfiler_4.14.0
[133] DOSE_4.0.0 tidytree_0.4.6 viridisLite_0.4.2
[136] sigora_3.1.1 scales_1.3.0 purrr_1.0.2
[139] crayon_1.5.3 GetoptLong_1.0.5 rlang_1.1.4
[142] cowplot_1.1.3 fastmatch_1.1-4 KEGGREST_1.46.0
参考
- https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012422
相关文章:
【R包】pathlinkR转录组数据分析和可视化利器
介绍 通常情况下,基因表达研究如微阵列和RNA-Seq会产生数百到数千个差异表达基因(deg)。理解如此庞大的数据集的生物学意义变得非常困难,尤其是在分析多个条件和比较的情况下。该软件包利用途径富集和蛋白-蛋白相互作用网络&…...

RPA 与 AI 结合:开启智能自动化新时代
RPA 与 AI 结合:开启智能自动化新时代 在当今数字化快速发展的时代,企业面临着海量的数据处理和复杂的业务流程,如何提高效率、降低成本、优化业务流程成为了企业关注的焦点。而 RPA(Robotic Process Automation,机器…...

[免费]Springboot+Vue在线文档管理系统【论文+源码+SQL脚本】
大家好,我是java1234_小锋老师,看到一个不错的SpringbootVue在线文档管理系统,分享下哈。 项目视频演示 【免费】SpringBootVue在线文档管理系统 Java毕业设计_哔哩哔哩_bilibili 项目介绍 随着科学技术的飞速发展,社会的方方面…...

内容选题与商业布局
内容选题与商业布局 提示词 1:人群链(User Chain)提示词 2:需求链(Demand Chain)提示词 3:环境 需求 优势 三要素提示词 4:垂直于“人性”而非只“赛道”提示词 5:基于…...

文字描边实现内黄外绿效果
网页使用 <!DOCTYPE html> <html> <head> <style> .text-effect {color: #ffd700; /* 黄色文字 */-webkit-text-stroke: 2px #008000; /* 绿色描边(兼容Webkit内核) */text-stroke: 2px #008000; /* 标准语法 *…...

网络协议 HTTP、HTTPS、HTTP/1.1、HTTP/2 对比分析
1. 基本定义 HTTP(HyperText Transfer Protocol) 应用层协议,用于客户端与服务器之间的数据传输(默认端口 80)。 HTTP/1.0:早期版本,每个请求需单独建立 TCP 连接,效率低。HTTP/1.1&…...

千峰React:Hooks(上)
什么是Hooks ref引用值 普通变量的改变一般是不好触发函数组件的渲染的,如果想让一般的数据也可以得到状态的保存,可以使用ref import { useState ,useRef} from reactfunction App() {const [count, setCount] useState(0)let num useRef(0)const h…...

salesforce 为什么无法关闭task,显示:insufficient access rights on object id
在 Salesforce 中,如果你在尝试关闭任务(Task)时遇到 “Insufficient access rights on object id” 错误,通常是由于以下几种可能的权限问题导致的: 1. 任务的所有权问题 Salesforce 中的任务(Task&…...

机器学习:强化学习的epsilon贪心算法
强化学习(Reinforcement Learning, RL)是一种机器学习方法,旨在通过与环境交互,使智能体(Agent)学习如何采取最优行动,以最大化某种累积奖励。它与监督学习和无监督学习不同,强调试错…...

MongoDB—(一主、一从、一仲裁)副本集搭建
MongoDB集群介绍: MongoDB 副本集是由多个MongoDB实例组成的集群,其中包含一个主节点(Primary)和多个从节点(Secondary),用于提供数据冗余和高可用性。以下是搭建 MongoDB 副本集的详细步骤&am…...

MyBatis TypeHandler 详解与实战:FastJson 实现字符串转 List
在 MyBatis 中,TypeHandler 是实现 Java 类型与数据库类型双向转换 的核心组件。无论是处理基础数据类型还是复杂的 JSON、枚举或自定义对象,它都能通过灵活的扩展机制满足开发需求。本文将通过一个 将数据库 JSON 字符串转换为 List<User> 的案例…...
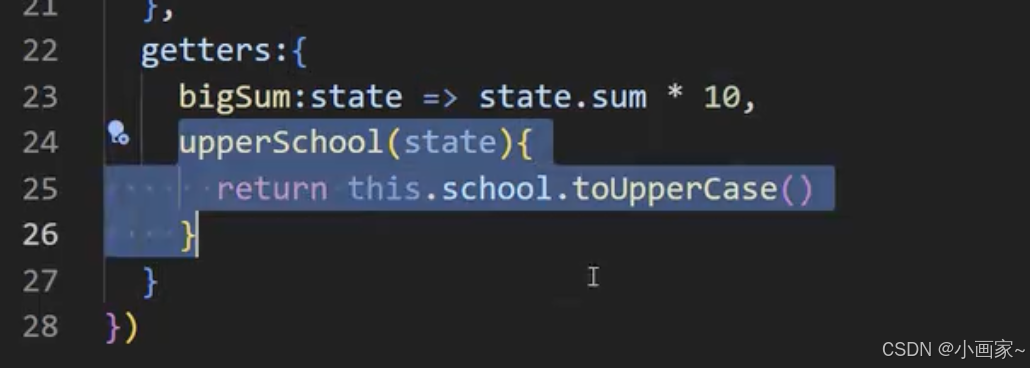
第二十八:5.5.【storeToRefs】5.6.【getters】
LoveTalk.vue: 调用: // 方法 const talkStore useTalkStore() function getLoveTalk(){ talkStore.getATalk() } 如果是要简短的形式调用: const talkStore useTalkStore() // user hooks 的形式调用 const {schoole,local} talkStore // …...

APISIX Dashboard上的配置操作
文章目录 登录配置路由配置消费者创建后端服务项目配置上游再创建一个路由测试 登录 http://192.168.10.101:9000/user/login?redirect%2Fdashboard 根据docker 容器里的指定端口: 配置路由 通过apisix 的API管理接口来创建(此路由,直接…...

MinIO在 Docker中修改登录账号和密码
MinIO在 Docker中修改登录账号和密码 随着云计算和大数据技术的快速发展,对象存储服务逐渐成为企业数据管理的重要组成部分。MinIO 作为一种高性能、分布式的对象存储系统,因其简单易用、高效可靠的特点而备受开发者青睐。然而,在实际应用中…...

英文论文查重,Turnitin和IThenticate两个系统哪个更合适?
Turnitin系统和IThenticate系统都是检测英文论文的查重系统,但是两者之间还是有一些不一样的。 下面针对这两个系统给大家具体分析一下。 一、Turnitin系统 Turnitin检测系统: https://truth-turnitin.similarity-check.com Turnitin是世界上主流的…...

pnpm的基本用法
以下是 pnpm 的核心命令和使用指南,涵盖从安装依赖到项目管理的常见操作: 1. 基础命令 (1) 安装依赖 pnpm install # 安装 package.json 中的所有依赖 pnpm install <包名> # 安装指定包(自动添加到 dependencies…...

【实战中提升自己】防火墙篇之双ISP切换与VRRP切换对于用户的体验
! 拓扑与说明 某公司的网络架构,这样的架构在目前的网络中是在常见的,假设您接收一个这样的网络,应该如何部署,该实战系列,就是一步一步讲解,如何规划、设计、部署这样一个环境,这…...

Go在1.22版本修复for循环陷阱
记录 前段时间升级Go版本碰到一个大坑,先记录。 先上代码案例: func main() {testClosure() }func testClosure() {for i : 0; i < 5; i {defer func() {fmt.Println(i)}()} }在1.22之下(不包括1.22)版本: 输出的…...

Nginx+PHP+MYSQL-Ubuntu在线安装
在 Ubuntu 上配置 Nginx、PHP 和 MySQL 的步骤如下: 1. 更新系统包 首先,确保系统包是最新的: sudo apt update sudo apt upgrade2. 安装 Nginx 安装 Nginx: sudo apt install nginx启动并启用 Nginx 服务: sudo…...

SpringDataJPA使用deleteAllInBatch方法逻辑删除失效
概述 在使用Spring Boot JPA时,执行批量删除操作时,遇到逻辑删除失效的问题。具体而言,当使用deleteAllInBatch方法时,数据会被物理删除,而不是进行逻辑删除;但是当使用deleteAll时,逻辑删除操…...
)
从STM32迁移到普冉PY32F003:UART代码移植保姆级教程(附HAL库对比)
从STM32到普冉PY32F003的UART代码迁移实战指南 1. 国产MCU替代浪潮下的技术选择 近年来,半导体行业的供应链波动促使更多工程师将目光投向国产MCU解决方案。普冉PY32F003系列作为Cortex-M0内核的代表产品,以48MHz主频、64KB Flash和8KB RAM的配置&#x…...

告别拍脑袋规划!用ArcGIS做绿道选线:如何科学量化坡度、水域、道路成本并加权计算
科学规划绿道的ArcGIS高阶技法:从成本栅格构建到最优路径生成绿道规划从来不是简单的"两点之间直线最短",而是需要综合考虑地形、生态、人文等多维因素的复杂决策过程。传统规划中常见的"拍脑袋"决策方式,往往导致建成后…...

对称与负电源测试:动态直流电子负载的设计、原理与应用
1. 项目概述:对称与负电源的静态与动态直流负载在电子实验室里,测试一个电源的性能,尤其是它的动态响应能力,是件既基础又关键的事。我们常说的“直流电子负载”就是这个领域的核心工具。我之前设计并分享过一个用于正电源测试的静…...

2026年,揭秘那些真正安全的原生态食材厂家你不可不知的秘密
随着人们生活水平的提升以及对健康的日益重视,选择真正安全的原生态食材已经成为许多人购买食物的标准。但市场的繁杂使得甄别真正安全的食材厂家变得愈加困难。今天,我将通过几个关键角度,为大家揭秘那些真正安全的原生态食材厂家的秘密&…...

Qri高级功能:如何使用JSON Schema验证和描述数据集结构
Qri高级功能:如何使用JSON Schema验证和描述数据集结构 【免费下载链接】qri youre invited to a data party! 项目地址: https://gitcode.com/gh_mirrors/qr/qri Qri是一个强大的开源数据协作工具,它提供了丰富的功能来帮助用户管理、共享和验证…...

16个分片+2副本:pg_shard的master_create_worker_shards最佳实践
16个分片2副本:pg_shard的master_create_worker_shards最佳实践 【免费下载链接】pg_shard ATTENTION: pg_shard is superseded by Citus, its more powerful replacement 项目地址: https://gitcode.com/gh_mirrors/pg/pg_shard pg_shard作为PostgreSQL的分…...

百度深度学习研究院的“叛将“,带着一颗芯片改变了中国智能驾驶——地平线余凯,从ImageNet冠军到征程出货1000万
大家好,我是写代码的篮球球痴。这篇文章跟我自己有点关系——我开的是理想汽车。理想的智驾系统 AD Pro,搭载的就是地平线征程 5 芯片。2026 年 1 月理想 AD Pro 4.0 推送,基于单颗征程 6M 实现了城市 NOA——这是行业里第一个用单颗 128TOPS…...

DeepSeek重复代码识别失效了?5个被90%团队忽略的AST解析盲区及修复清单
更多请点击: https://codechina.net 第一章:DeepSeek代码重复检测失效的真相与影响 DeepSeek-R1 模型在代码理解任务中表现出色,但其内置的代码重复检测机制在特定场景下存在系统性失效。根本原因在于模型对语义等价但语法结构差异显著的代…...

Log4Shell漏洞深度解析:Spring Boot日志注入原理与四层修复方案
1. 这个漏洞不是“远程执行代码”那么简单——它是一次对Java生态信任链的系统性击穿Log4j CVE-2021-44228,业内常简称为“Log4Shell”,2021年12月爆发时,我正在给一家金融客户的Spring Boot微服务集群做灰度发布前的安全加固。凌晨三点收到告…...

别再把大模型当搜索框了:一文讲透 LLM 的基本原理、能力边界与局限性
写在前面很多人把大语言模型当成“会聊天的搜索引擎”,结果一上线就遇到幻觉、口径不稳、上下文丢失、成本失控。真正理解 LLM,要先抓住一句话:它是基于 Transformer 的概率生成模型,核心能力来自海量预训练、上下文学习与后训练对…...