基于qiime2的16S数据分析全流程:从导入数据到下游分析一条龙
目录
创建metadata
把数据导入qiime2
去除引物序列
双端合并 (dada2不需要)
质控 (dada2不需要)
使用deblur获得特征序列
使用dada2生成代表序列与特征表
物种鉴定
可视化物种鉴定结果
构建进化树(ITS一般不构建进化树)
生成多样性分析所需数据
α多样性分析
beta多样性分析
绘制稀释曲线
差异物种分析
如何把.qza格式文件导出?比如把特征表导出
演示单端数据导入qiime2


创建metadata


把数据导入qiime2
去除引物序列
双端合并 (dada2不需要)
质控 (dada2不需要)
使用deblur获得特征序列

使用dada2生成代表序列与特征表
物种鉴定
可视化物种鉴定结果


构建进化树(ITS一般不构建进化树)


生成多样性分析所需数据


α多样性分析

beta多样性分析

绘制稀释曲线

差异物种分析
如何把.qza格式文件导出?比如把特征表导出
演示单端数据导入qiime2

相关文章:
基于qiime2的16S数据分析全流程:从导入数据到下游分析一条龙
目录 创建metadata 把数据导入qiime2 去除引物序列 双端合并 (dada2不需要) 质控 (dada2不需要) 使用deblur获得特征序列 使用dada2生成代表序列与特征表 物种鉴定 可视化物种鉴定结果 构建进化树(ITS一般不构建进化树…...
)
【软件测试开发】:软件测试常用函数1.0(C++)
1. 元素的定位 web⾃动化测试的操作核⼼是能够找到⻚⾯对应的元素,然后才能对元素进⾏具体的操作。 常⻅的元素定位⽅式⾮常多,如id,classname,tagname,xpath,cssSelector 常⽤的主要由cssSelector和xpath…...

vue2项目修改浏览器显示的网页图标
1.准备一个新的图标文件,通常是. ico格式,也可以是. Png、. Svg等格式 2.将新的图标文件(例如:faviconAt.png)放入项目的public文件夹中。如下图 public文件夹中的所有文件都会在构建时原样复制到最终的输出目录(通常是dist) 3. 修改vue项目…...

开源、创新与人才发展:机器人产业的战略布局与稚晖君成功案例解析
目录 引言 一、开源:机器人产业的战略布局 促进技术进步和生态建设 吸引人才和合作伙伴 建立标准和网络效应 降低研发风险与成本 二、稚晖君:华为"天才少年计划"的成功典范 深厚的技术积累与动手能力 强烈的探索和创新意识 持续公开…...

线程相关作业
1.创建两个线程,分支线程1拷贝文件的前一部分,分支线程2拷贝文件的后一部分 #include "head.h"#define BUFFER_SIZE 1024// 线程参数结构体,包含文件名和文件偏移量 typedef struct {FILE *src_file;FILE *dest_file;long start_o…...

通义万相2.1开源版本地化部署攻略,生成视频再填利器
2025 年 2 月 25 日晚上 11:00 通义万相 2.1 开源发布,前两周太忙没空搞它,这个周末,也来本地化部署一个,体验生成效果如何,总的来说,它在国内文生视频、图生视频的行列处于领先位置,…...

【模拟CMOS集成电路设计】带隙基准(Bandgap)设计与仿真(基于运放的电流模BGR)
【模拟CMOS集成电路设计】带隙基准(Bandgap)设计与仿真 前言工程文件&部分参数计算过程,私聊~ 一、 设计指标指标分析: 二、 电路分析三、 仿真3.1仿真电路图3.2仿真结果(1)运放增益(2)基准温度系数仿真(3)瞬态启动仿真(4)静态…...

如何选择国产串口屏?
目录 1、迪文 2、淘晶驰 3、广州大彩 4、金玺智控 5、欣瑞达 6、富莱新 7、冠显 8、有彩 串口屏,顾名思义,就是通过串口通信接口(如RS232、RS485、TTL UART等)与主控设备进行通信的显示屏。其核心功能是显示信息和接收输入…...

Solana中的程序派生地址(PDAs):是什么,为什么,以及如何?
程序派生地址 (PDA) 在 Solana 中的应用:什么、为什么和如何? 在学习 Solana 时,你会经常听到关于 程序派生地址 (PDAs) 的讨论。它们就像这样 —— 强大、多功能,而且最重要的是,稍微被误解。如果你是一个开发者&…...

利用FatJar彻底解决Jar包冲突(一)
利用FatJar彻底解决Jar包冲突 序FatJar的加载与隔离⼀、 FatJar概念⼆、FatJar的加载三、FatJar的隔离四、隔离机制验证五、 FatJar的定位六、 打包注意点 序 今天整理旧电脑里的资料,偶然翻到大概10年前实习时写的笔记,之前经常遇到Java依赖冲突的问题…...

Spring MVC笔记
01 什么是Spring MVC Spring MVC 是 Spring 框架中的一个核心模块,专门用于构建 Web 应用程序。它基于经典的 MVC 设计模式(Model-View-Controller),但通过 Spring 的特性(如依赖注入、注解驱动)大幅简化了…...
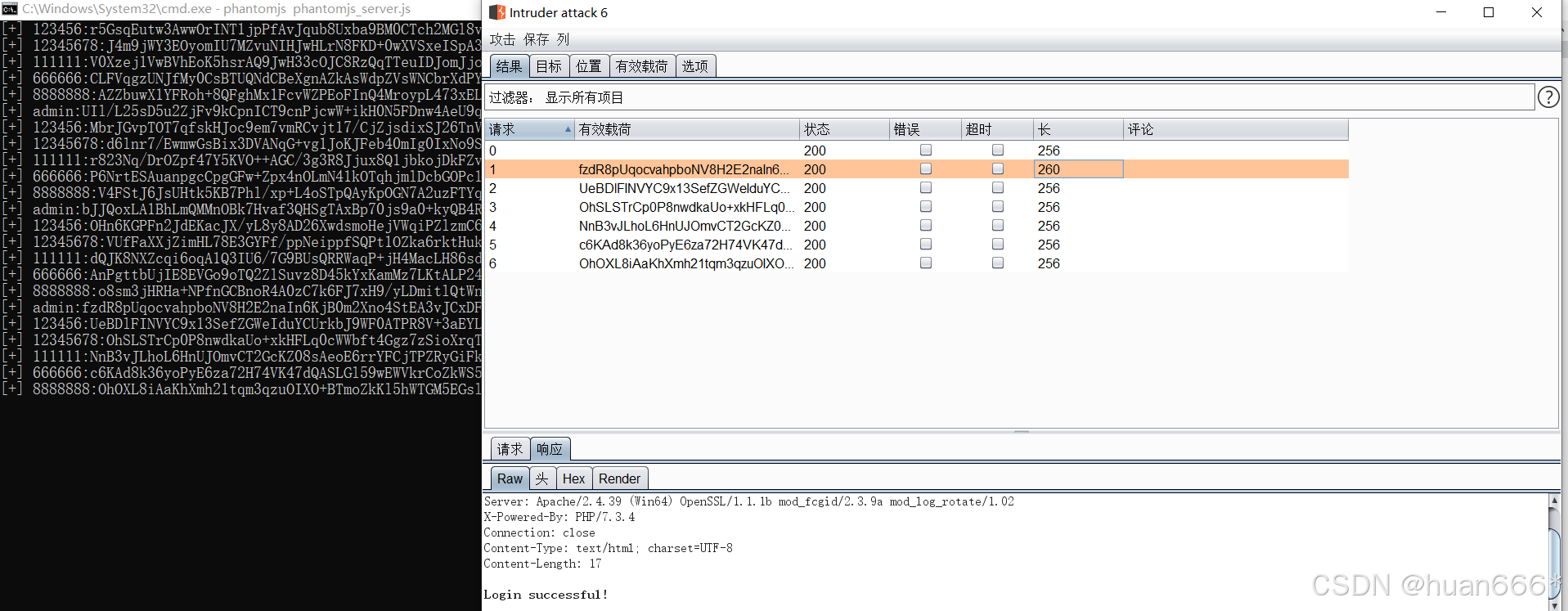
BurpSuite插件jsEncrypter使用教程
一、前言 在当今Web应用安全测试中,前端加密已成为开发者保护敏感数据的常用手段。然而,这也给安全测试人员带来了挑战,传统的抓包方式难以获取明文数据,测试效率大打折扣。BurpSuite作为一款强大的Web安全测试工具,其…...

【C#实现手写Ollama服务交互,实现本地模型对话】
前言 C#手写Ollama服务交互,实现本地模型对话 最近使用C#调用OllamaSharpe库实现Ollama本地对话,然后思考着能否自己实现这个功能。经过一番查找,和查看OllamaSharpe源码发现确实可以。其实就是开启Ollama服务后,发送HTTP请求&a…...

Android Glide 框架线程管理模块原理的源码级别深入分析
一、引言 在现代的 Android 应用开发中,图片加载是一个常见且重要的功能。Glide 作为一款广泛使用的图片加载框架,以其高效、灵活和易用的特点受到了开发者的青睐。其中,线程管理模块是 Glide 框架中至关重要的一部分,它负责协调…...

每天记录一道Java面试题---day32
MySQL索引的数据结构、各自优劣 回答重点 B树:是一个平衡的多叉树,从根节点到每个叶子节点的高度差不超过1,而且同层级的节点间有指针相互连接。在B树上的常规检索,从根节点到叶子节点的搜索效率基本相当,不会出现大…...

Vue3 Pinia 符合直觉的Vue.js状态管理库
Pinia 符合直觉的Vue.js状态管理库 什么时候使用Pinia 当两个关系非常远的组件,要传递参数时使用Pinia组件的公共参数使用Pinia...

深度学习与大模型基础-向量
大家好!今天我们来聊聊向量(Vector)。别被这个词吓到,其实向量在我们的生活中无处不在,只是我们没注意罢了。 1. 向量是什么? 简单来说,向量就是有大小和方向的量。比如你从家走到学校&#x…...

【网络编程】完成端口 IOCP
10.11 完成端口 10.11.1 基本概念 完成端口的全称是I/O 完成端口,英文为IOCP(I/O Completion Port) 。IOCP是一个异 步I/O 的 API, 可以高效地将I/O 事件通知给应用程序。与使用select() 或是其他异步方法不同 的是,一个套接字与一个完成端口关联了起来…...

《苍穹外卖》SpringBoot后端开发项目重点知识整理(DAY1 to DAY3)
目录 一、在本地部署并启动Nginx服务1. 解压Nginx压缩包2. 启动Nginx服务3. 验证Nginx是否启动成功: 二、导入接口文档1. 黑马程序员提供的YApi平台2. YApi Pro平台3. 推荐工具:Apifox 三、Swagger1. 常用注解1.1 Api与ApiModel1.2 ApiModelProperty与Ap…...

管理网络安全
防火墙在 Linux 系统安全中有哪些重要的作用? 防火墙作为网络安全的第一道防线,能够根据预设的规则,对进出系统的网络流量进行严格筛选。它可以阻止未经授权的外部访问,只允许符合规则的流量进入系统,从而保护系统免受…...

EurekaClaw:多智能体AI研究助手,自动化实现从灵感到论文的完整流程
1. 项目概述:从灵感到论文的自动化研究助手在科研工作中,最令人兴奋又最耗费精力的,莫过于从零散的文献、模糊的直觉中,一步步构建出严谨的、可发表的成果。这个过程通常需要经历文献调研、假设生成、理论证明、实验验证和论文撰写…...

从游戏角色到人脸分析:聊聊‘摇头、点头、转头’背后的欧拉角与万向节死锁
游戏角色控制与人脸分析的奇妙交汇:解码欧拉角与万向节死锁 想象一下你在玩一款3A级开放世界游戏:按下左摇杆,角色开始左右张望;推动右摇杆,角色抬头望向天空中的飞龙;同时扳动两个摇杆,角色做出…...

LLM训练实战:8个编程谜题带你掌握分布式训练核心技术
1. 项目概述与核心价值如果你对大型语言模型(LLM)的训练过程感到好奇,或者你听说过“千卡集群”、“万亿参数”这些词,但总觉得它们离自己很遥远,那么这个名为“LLM Training Puzzles”的项目,就是为你量身…...

JSON数据高效处理:命令行工具jsoncut的查询、过滤与投影实战
1. 项目概述:一个专为JSON数据“瘦身”的利器在前后端开发、API接口调试、数据迁移或者日志分析的日常工作中,JSON格式的数据几乎无处不在。它结构清晰、易于阅读和解析,是现代数据交换的绝对主力。但随之而来的一个常见痛点就是:…...

ARM GICv5 ITS_CR1寄存器配置与中断优化实践
1. ARM GICv5 ITS架构概述中断控制器是现代计算机系统中的关键组件,负责管理和分发硬件中断请求。ARM GICv5架构中的Interrupt Translation Service (ITS)模块通过创新的设备ID和事件ID映射机制,实现了灵活高效的中断路由方案。ITS作为GICv5的可选扩展组…...

Godot引擎开发实战:高效利用代码食谱仓库加速游戏原型设计
1. 项目概述:一个为Godot开发者量身定制的“食谱”仓库如果你正在使用Godot引擎,无论是刚入门的新手,还是已经摸爬滚打了一段时间的开发者,大概率都经历过这样的时刻:脑子里有一个很酷的游戏机制想法,比如“…...

AI Agent Harness Engineering 未来生态:开源 vs 闭源的竞争与合作格局
AI Agent Harness Engineering 未来生态:开源 vs 闭源的竞争与合作格局 引言:AI Agent不是终点,Harness才是通用智能落地的核心阀门 1.1 从“AI大模型(LLM)元年”到“AI Agent生态元年”:技术拐点的悄然发…...

Arm嵌入式多线程编程:原理、实践与优化
1. Arm嵌入式开发中的多线程编程基础在嵌入式系统开发中,多线程编程是提高系统响应能力和资源利用率的重要手段。Arm架构作为嵌入式领域的主流处理器架构,其编译器工具链对多线程编程提供了完善的支持。不同于通用计算环境,嵌入式系统的多线程…...

数据库测试的盲区:用AI生成边界值,发现隐藏的数据异常
在软件测试领域,数据库层的质量保障常常陷入一种“平静的假象”——核心CRUD操作通过、索引命中率达标、慢查询被优化,一切看似井然有序。然而线上事故统计却揭示了一个残酷的事实:超过七成的数据库相关故障并非源于架构缺陷或性能瓶颈&#…...

云原生 Kubernetes 核心概念与组件详解
目录 一、Kubernetes 是什么? 核心功能概览 二、部署演进:从物理机到容器 1. 传统部署时代 2. 虚拟化部署时代 3. 容器部署时代 三、Kubernetes 集群架构 1. 控制平面组件(集群大脑) (1)kube-apise…...