文章MSM_metagenomics(三):Alpha多样性分析
欢迎大家关注全网生信学习者系列:
- WX公zhong号:生信学习者
- Xiao hong书:生信学习者
- 知hu:生信学习者
- CDSN:生信学习者2
介绍
本教程使用基于R的函数来估计微生物群落的香农指数和丰富度,使用MetaPhlAn profile数据。估计结果进一步进行了可视化,并与元数据关联,以测试统计显著性。
数据
大家通过以下链接下载数据:
- 百度网盘链接:https://pan.baidu.com/s/1f1SyyvRfpNVO3sLYEblz1A
- 提取码: 请关注WX公zhong号_生信学习者_后台发送 复现msm 获取提取码
R 包
- SummarizedExperiment
- mia
- ggpubr
- ggplot2
- lfe
Alpha diversity estimation and visualization
使用alpha_diversity_funcs.R计算alpha多样性和可视化。
- 代码
SE_converter <- function(md_rows, tax_starting_row, mpa_md) {# SE_converter function is to convery metadata-wedged mpa table into SummarisedExperiment structure.# md_rows: a vector specifying the range of rows indicating metadata.# tax_starting_row: an interger corresponding to the row where taxonomic abundances start.# mpa_md: a metaphlan table wedged with metadata, in the form of dataframe.md_df <- mpa_md[md_rows,] # extract metadata part from mpa_md tabletax_df <- mpa_md[tax_starting_row: nrow(mpa_md),] # extract taxonomic abundances part from mpa_md table### convert md_df to a form compatible with SummarisedExperiment ### SE_md_df <- md_df[, -1]rownames(SE_md_df) <- md_df[, 1]SE_md_df <- t(SE_md_df)### convert md_df to a form compatible with SummarisedExperiment ###### prep relab values in a form compatible with SummarisedExperiment ###SE_relab_df <- tax_df[, -1]rownames(SE_relab_df) <- tax_df[, 1]col_names <- colnames(SE_relab_df)SE_relab_df[, col_names] <- apply(SE_relab_df[, col_names], 2, function(x) as.numeric(as.character(x)))### prep relab values in a form compatible with SummarisedExperiment ###SE_tax_df <- tax_df[, 1:2]rownames(SE_tax_df) <- tax_df[, 1]SE_tax_df <- SE_tax_df[-2]colnames(SE_tax_df) <- c("species")SE_data <- SummarizedExperiment::SummarizedExperiment(assays = list(relative_abundance = SE_relab_df),colData = SE_md_df,rowData = SE_tax_df)SE_data
}est_alpha_diversity <- function(se_data) {# This function is to estimate alpha diversity (shannon index and richness) of a microbiome and output results in dataframe.# se_data: the SummarizedExperiment data structure containing metadata and abundance values.se_data <- se_data |>mia::estimateRichness(abund_values = "relative_abundance", index = "observed")se_data <- se_data |>mia::estimateDiversity(abund_values = "relative_abundance", index = "shannon")se_alpha_div <- data.frame(SummarizedExperiment::colData(se_data))se_alpha_div
}make_boxplot <- function(df, xlabel, ylabel, font_size = 11, jitter_width = 0.2, dot_size = 1, font_style = "Arial", stats = TRUE, pal = NULL) {# This function is to create a boxplot using categorical data.# df: The dataframe containing microbiome alpha diversities, e.g. `shannon` and `observed` with categorical metadata.# xlabel: the column name one will put along x-axis.# ylabel: the index estimate one will put along y-axis.# font_size: the font size, default: [11]# jitter_width: the jitter width, default: [0.2]# dot_size: the dot size inside the boxplot, default: [1]# font_style: the font style, default: `Arial`# pal: a list of color codes for pallete, e.g. c(#888888, #eb2525). The order corresponds the column order of boxplot.# stats: wilcox rank-sum test. default: TRUEif (stats) {nr_group = length(unique(df[, xlabel])) # get the number of groupsif (nr_group == 2) {group_pair = list(unique(df[, xlabel]))ggpubr::ggboxplot(data = df, x = xlabel, y = ylabel, color = xlabel,palette = pal, ylab = ylabel, xlab = xlabel,add = "jitter", add.params = list(size = dot_size, jitter = jitter_width)) +ggpubr::stat_compare_means(comparisons = group_pair, exact = T, alternative = "less") +ggplot2::stat_summary(fun.data = function(x) data.frame(y = max(df[, ylabel]), label = paste("Mean=",mean(x))), geom="text") +ggplot2::theme(text = ggplot2::element_text(size = font_size, family = font_style))} else {group_pairs = my_combn(unique((df[, xlabel])))ggpubr::ggboxplot(data = df, x = xlabel, y = ylabel, color = xlabel,palette = pal, ylab = ylabel, xlab = xlabel,add = "jitter", add.params = list(size = dot_size, jitter = jitter_width)) +ggpubr::stat_compare_means() + ggpubr::stat_compare_means(comparisons = group_pairs, exact = T, alternative = "greater") +ggplot2::stat_summary(fun.data = function(x) data.frame(y= max(df[, ylabel]), label = paste("Mean=",mean(x))), geom="text") +ggplot2::theme(text = ggplot2::element_text(size = font_size, family = font_style))}} else {ggpubr::ggboxplot(data = df, x = xlabel, y = ylabel, color = xlabel,palette = pal, ylab = ylabel, xlab = xlabel,add = "jitter", add.params = list(size = dot_size, jitter = jitter_width)) +ggplot2::theme(text = ggplot2::element_text(size = font_size, family = font_style))}
}my_combn <- function(x) {combs <- list()comb_matrix <- combn(x, 2)for (i in 1: ncol(comb_matrix)) {combs[[i]] <- comb_matrix[,i]}combs
}felm_fixed <- function(data_frame, f_factors, t_factor, measure) {# This function is to perform fixed effect linear modeling# data_frame: a dataframe containing measures and corresponding effects # f_factors: a vector of header names in the dataframe which represent fixed effects# t_factors: test factor name in the form of string# measure: the measured values in column, e.g., shannon or richness
# all_factors <- c(t_factor, f_factors)
# for (i in all_factors) {
# vars <- unique(data_frame[, i])
# lookup <- setNames(seq_along(vars) -1, vars)
# data_frame[, i] <- lookup[data_frame[, i]]
# }
# View(data_frame)str1 <- paste0(c(t_factor, paste0(f_factors, collapse = " + ")), collapse = " + ")str2 <- paste0(c(measure, str1), collapse = " ~ ")felm_stats <- lfe::felm(eval(parse(text = str2)), data = data_frame)felm_stats
}
加载一个包含元数据和分类群丰度的合并MetaPhlAn profile文件
mpa_df <- data.frame(read.csv("./data/merged_abundance_table_species_sgb_md.tsv", header = TRUE, sep = "\t"))
| sample | P057 | P054 | P052 | … | P049 |
|---|---|---|---|---|---|
| sexual_orientation | MSM | MSM | MSM | … | Non-MSM |
| HIV_status | negative | positive | positive | … | negative |
| ethnicity | Caucasian | Caucasian | Caucasian | … | Caucasian |
| antibiotics_6month | Yes | No | No | … | No |
| BMI_kg_m2_WHO | ObeseClassI | Overweight | Normal | … | Overweight |
| Methanomassiliicoccales_archaeon | 0.0 | 0.0 | 0.0 | … | 0.01322 |
| … | … | … | … | … | … |
| Methanobrevibacter_smithii | 0.0 | 0.0 | 0.0 | … | 0.19154 |
接下来,我们将数据框转换为SummarizedExperiment数据结构,以便使用SE_converter函数继续分析,该函数需要指定三个参数:
md_rows: a vector specifying the range of rows indicating metadata. Note: 1-based.tax_starting_row: an interger corresponding to the row where taxonomic abundances start.mpa_md: a metaphlan table wedged with metadata, in the form of dataframe.
SE <- SE_converter(md_rows = 1:5,tax_starting_row = 6, mpa_md = mpa_df)SE class: SummarizedExperiment
dim: 1676 139
metadata(0):
assays(1): relative_abundance
rownames(1676): Methanomassiliicoccales_archaeon|t__SGB376GGB1567_SGB2154|t__SGB2154 ... Entamoeba_dispar|t__EUK46681Blastocystis_sp_subtype_1|t__EUK944036
rowData names(1): species
colnames(139): P057 P054 ... KHK16 KHK11
colData names(5): sexual_orientation HIV_status ethnicityantibiotics_6month BMI_kg_m2_WHO
接下来,我们可以使用est_alpha_diversity函数来估计每个宏基因组样本的香农指数和丰富度。
alpha_df <- est_alpha_diversity(se_data = SE)
alpha_df
| sexual_orientation | HIV_status | ethnicity | antibiotics_6month | BMI_kg_m2_WHO | observed | shannon | |
|---|---|---|---|---|---|---|---|
| P057 | MSM | negative | Caucasian | Yes | ObeseClassI | 134 | 3.1847 |
| P054 | MSM | positive | Caucasian | No | Overweight | 141 | 2.1197 |
| … | … | … | … | … | … | … | … |
| P052 | MSM | positive | Caucasian | No | Normal | 152 | 2.5273 |
为了比较不同组之间的alpha多样性差异,我们可以使用make_boxplot函数,并使用参数:
df: The dataframe containing microbiome alpha diversities, e.g.shannonandobservedwith categorical metadata.xlabel: the column name one will put along x-axis.ylabel: the index estimate one will put along y-axis.font_size: the font size, default: [11]jitter_width: the jitter width, default: [0.2]dot_size: the dot size inside the boxplot, default: [1]font_style: the font style, default:Arialpal: a list of color codes for pallete, e.g. c(#888888, #eb2525). The order corresponds the column order of boxplot.stats: wilcox rank-sum test. default:TRUE
shannon <- make_boxplot(df = alpha_df,xlabel = "sexual_orientation",ylabel = "shannon",stats = TRUE,pal = c("#888888", "#eb2525"))richness <- make_boxplot(df = alpha_df,xlabel = "sexual_orientation", ylabel = "observed",stats = TRUE,pal = c("#888888", "#eb2525"))
multi_plot <- ggpubr::ggarrange(shannon, richness, ncol = 2)
ggplot2::ggsave(file = "shannon_richness.svg", plot = multi_plot, width = 4, height = 5)
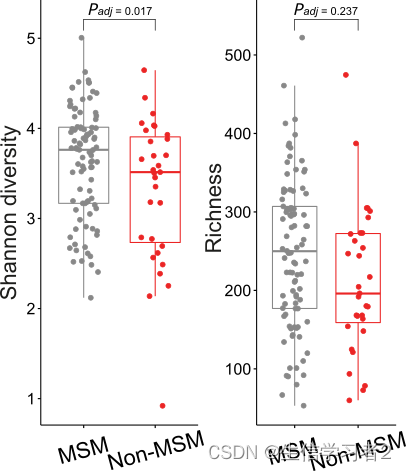
通过固定效应线性模型估计关联的显著性
在宏基因组分析中,除了感兴趣的变量(例如性取向)之外,通常还需要处理多个变量(例如HIV感染和抗生素使用)。因此,在测试微生物群落矩阵(例如香农指数或丰富度)与感兴趣的变量(例如性取向)之间的关联时,控制这些混杂效应非常重要。在这里,我们使用基于固定效应线性模型的felm_fixed函数,该函数实现在R包lfe 中,以估计微生物群落与感兴趣变量之间的关联显著性,同时控制其他变量的混杂效应。
data_frame: The dataframe containing microbiome alpha diversities, e.g.shannonandobservedwith multiple variables.f_factors: A vector of variables representing fixed effects.t_factor: The variable of interest for testing.measure: The header indicating microbiome measure, e.g.shannonorrichness
lfe_stats <- felm_fixed(data_frame = alpha_df,f_factors = c(c("HIV_status", "antibiotics_6month")),t_factor = "sexual_orientation",measure = "shannon")
summary(lfe_stats)
Residuals:Min 1Q Median 3Q Max
-2.3112 -0.4666 0.1412 0.5200 1.4137 Coefficients:Estimate Std. Error t value Pr(>|t|)
(Intercept) 3.62027 0.70476 5.137 9.64e-07 ***
sexual_orientationMSM 0.29175 0.13733 2.125 0.0355 *
HIV_statuspositive -0.28400 0.14658 -1.937 0.0548 .
antibiotics_6monthNo -0.10405 0.67931 -0.153 0.8785
antibiotics_6monthYes 0.01197 0.68483 0.017 0.9861
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1Residual standard error: 0.6745 on 134 degrees of freedom
Multiple R-squared(full model): 0.07784 Adjusted R-squared: 0.05032
Multiple R-squared(proj model): 0.07784 Adjusted R-squared: 0.05032
F-statistic(full model):2.828 on 4 and 134 DF, p-value: 0.02725
F-statistic(proj model): 2.828 on 4 and 134 DF, p-value: 0.02725
相关文章:
文章MSM_metagenomics(三):Alpha多样性分析
欢迎大家关注全网生信学习者系列: WX公zhong号:生信学习者Xiao hong书:生信学习者知hu:生信学习者CDSN:生信学习者2 介绍 本教程使用基于R的函数来估计微生物群落的香农指数和丰富度,使用MetaPhlAn prof…...

Web前端与其他前端:深度对比与差异性剖析
Web前端与其他前端:深度对比与差异性剖析 在快速发展的前端技术领域,Web前端无疑是其中最耀眼的明星。然而,当我们谈论前端时,是否仅仅指的是Web前端?实际上,前端技术还包括了许多其他细分领域。本文将从四…...

AI 客服定制:LangChain集成订单能力
为了提高AI客服的问题解决能力,我们引入了LangChain自定义能力,并集成了订单能力。这使得AI客服可以根据用户提出的问题,自动调用订单接口,获取订单信息,并结合文本知识库内容进行回答。这种能力的应用,使得…...
【计算机毕业设计】242基于微信小程序的外卖点餐系统
🙊作者简介:拥有多年开发工作经验,分享技术代码帮助学生学习,独立完成自己的项目或者毕业设计。 代码可以私聊博主获取。🌹赠送计算机毕业设计600个选题excel文件,帮助大学选题。赠送开题报告模板ÿ…...

java程序监控linux服务器硬件,cpu、mem、disk等
实现 使用Oshi和Hutool工具包1、pom依赖<dependency><groupId>org.springframework.boot</groupId><artifactId>spring-boot-starter-web</artifactId></dependency><dependency><groupId>com.github.oshi</groupId>&l…...

高考报志愿闲谈
当你的朋友在选择大学和专业时寻求建议,作为一名研究生并有高考经验的人,你可以提供一些有价值的见解和建议。 兴趣与职业目标: 首先询问他对哪些工科领域感兴趣,如机械工程、电子工程、计算机科学等。探讨他的职业目标。了解他将…...

面试官考我Object类中的所有方法及场景使用?我...
咦咦咦,各位小可爱,我是你们的好伙伴——bug菌,今天又来给大家普及Java 知识点啦,别躲起来啊,听我讲干货还不快点赞,赞多了我就有动力讲得更嗨啦!所以呀,养成先点赞后阅读的好习惯&a…...

Web前端精通教程:深入探索与实战指南
Web前端精通教程:深入探索与实战指南 在数字化时代,Web前端技术已经成为构建优秀用户体验的基石。想要精通Web前端,不仅需要掌握扎实的基础知识,还需要具备丰富的实战经验和深入的思考。本文将从四个方面、五个方面、六个方面和七…...

四轴飞行器、无人机(STM32、NRF24L01)
一、简介 此电路由STM32为主控芯片,NRF24L01、MPU6050为辅,当接受到信号时,处理对应的指令。 二、实物图 三、部分代码 void FlightPidControl(float dt) { volatile static uint8_t statusWAITING_1; switch(status) { case WAITING_1: //等待解锁 if…...

OpenAI Assistants API:如何使用代码或无需代码创建您自己的AI助手
Its now easier than ever to create your own AI Assistant that can handle a lot of computing tasks for you. See how you can get started with the OpenAI AI Assistant API. 现在比以往任何时候都更容易创建您自己的AI助手,它可以为您处理许多计算任务。了…...
CentOS7 配置Nginx域名HTTPS
Configuring Nginx with HTTPS on CentOS 7 involves similar steps to the ones for Ubuntu, but with some variations in package management and service control. Here’s a step-by-step guide for CentOS 7: Prerequisites Domain Name: “www.xxx.com”Nginx Install…...

C++入门8 构造函数析构函数顺序|拷贝构造
一,构造函数析构函数 调用顺序 我们先来看下面的代码: #define _CRT_SECURE_NO_WARNINGS #include <iostream> #include <cstring> using namespace std; class student { public:char my_name[20];int my_id;student(int a) {my_id a;co…...

【git使用四】git分支理解与操作(详解)
目录 (1)理解git分支 主分支(主线) 功能分支 主线和分支关系 将分支合并到主分支 快速合并 非快速合并 git代码管理流程 (2)理解git提交对象 提交对象与commitID Git如何保存数据 示例讲解 &a…...
【docker】如何解决artalk的跨域访问问题
今天折腾halo的时候,发现artalk出现跨域访问报错,内容如下。 Access to fetch at https://artk.musnow.top/api/stat from origin https://halo.musnow.top has been blocked by CORS policy: The Access-Control-Allow-Origin header contains multipl…...
MYSQL 索引下推 45讲
刘老师群里,看到一位小友 问<MYSQL 45讲>林晓斌的回答 大意是一个组合索引 (a,b,c) 条件 a > 5 and a <10 and b123, 这样的情况下是如何? 林老师给的回答是 A>5 ,然后下推B123 小友 问 "为什么不是先 进行范围查询,然后在索引下推 b123?" 然后就…...
CentOS7服务器中安装openCV4.8的教程
参考链接:Centos7环境下cmake3.25的编译与安装 参考链接:Linux安装或者升级cmake,例子为v3.10.2升级到v3.25.0(自己指定版本) 参考链接:Linux安装Opencv(C) 一、下载资源 1.下载cmake3.25.0的压缩包&am…...
Java课程设计:基于swing的贪吃蛇小游戏
文章目录 一、项目介绍二、核心代码三、项目展示四、源码获取 一、项目介绍 贪吃蛇是一款经典的休闲益智游戏,自问世以来便深受广大用户的喜爱。这个游戏的基本玩法是控制一条不断增长的蛇,目标是吃掉屏幕上出现的食物,同时避免撞到边缘或自身。随着游戏的进行,蛇的身体会越长…...

【HarmonyOS】HUAWEI DevEco Studio 下载地址汇总
目录 OpenHarmony 4.x Releases 4.1 Release4.0 Release OpenHarmony 3.x Releases 3.2.1 Release3.2 Release3.1.3 Release3.1.2 Release3.1.1 Release3.1 Release 说明 Full SDK:面向OEM厂商提供,包含了需要使用系统权限的系统接口。 Public SDK&am…...
)
华为OD刷题C卷 - 每日刷题30(小明找位置,分隔均衡字符串)
1、(小明找位置): 这段代码是解决“小明找位置”的问题。它提供了一个Java类Main,其中包含main方法和getResult方法,用于帮助小明快速找到他在排队中应该站的位置。 main方法首先读取已排列好的小朋友的学号数组和小…...

SOFTS: 时间序列预测的最新模型以及Python使用示例
近年来,深度学习一直在时间序列预测中追赶着提升树模型,其中新的架构已经逐渐为最先进的性能设定了新的标准。 这一切都始于2020年的N-BEATS,然后是2022年的NHITS。2023年,PatchTST和TSMixer被提出,最近的iTransforme…...

Promises/A+性能优化指南:让你的异步代码运行得更快
Promises/A性能优化指南:让你的异步代码运行得更快 【免费下载链接】promises-spec An open standard for sound, interoperable JavaScript promises—by implementers, for implementers. 项目地址: https://gitcode.com/gh_mirrors/pr/promises-spec 在Ja…...

PlantUML Editor:用代码思维重塑UML绘图的现代工具
PlantUML Editor:用代码思维重塑UML绘图的现代工具 【免费下载链接】plantuml-editor PlantUML online demo client 项目地址: https://gitcode.com/gh_mirrors/pl/plantuml-editor 你是否厌倦了传统拖拽式UML工具的繁琐操作?PlantUML Editor将彻…...

MPLAB® Harmony嵌入式框架实战:从架构解析到项目开发避坑指南
1. 项目概述:从零到一,理解MPLAB Harmony的价值如果你是一位嵌入式开发者,尤其是长期与Microchip的PIC或SAM系列MCU打交道的朋友,那么“MPLAB Harmony”这个名字你一定不陌生。它可能出现在官方文档的角落里,在论坛的讨…...

对比直接使用原厂 API 体验 Taotoken 在模型选型上的便捷性
🚀 告别海外账号与网络限制!稳定直连全球优质大模型,限时半价接入中。 👉 点击领取海量免费额度 对比直接使用原厂 API 体验 Taotoken 在模型选型上的便捷性 当开发者需要评估不同大模型的能力以适配具体项目时,通常会…...

基于KB2040与Kailh大键的DIY宏键盘:从电路原理到3D打印全流程
1. 项目概述与核心思路 如果你和我一样,每天在电脑前要重复执行大量组合键操作,比如设计师频繁切换工具、程序员调试代码、视频剪辑师来回切时间轴,那么一个自定义的宏键盘绝对是效率神器。市面上的成品宏键盘要么键位固定,要么价…...

嵌入式音频处理与SD卡系统克隆实战指南
1. 项目概述与核心价值如果你正在捣鼓一块像Chumby Hacker Board这样的嵌入式开发板,或者任何带有音频输出和SD卡存储的Linux设备,那么你迟早会碰到两个绕不开的“硬骨头”:音频信号的处理和存储系统的克隆部署。前者决定了你的设备能不能“好…...

5个实战案例:使用Promises/A+规范解决复杂异步编程难题
5个实战案例:使用Promises/A规范解决复杂异步编程难题 【免费下载链接】promises-spec An open standard for sound, interoperable JavaScript promises—by implementers, for implementers. 项目地址: https://gitcode.com/gh_mirrors/pr/promises-spec P…...

3D模型格式转换终极指南:如何用stltostp快速将STL转为STEP格式
3D模型格式转换终极指南:如何用stltostp快速将STL转为STEP格式 【免费下载链接】stltostp Convert stl files to STEP brep files 项目地址: https://gitcode.com/gh_mirrors/st/stltostp 你是否曾经遇到这样的困境?辛苦设计的3D打印模型在STL格式…...

NewLife.Core配置系统深度解析:XML/JSON/HTTP多源配置实战
NewLife.Core配置系统深度解析:XML/JSON/HTTP多源配置实战 【免费下载链接】X Core basic components: log (file / network), configuration (XML / JSON / HTTP), cache (memory / redis), network (TCP / UDP / HTTP), RPC framework, serialization (binary / X…...

基于MCP协议构建AI驱动的加密货币数据智能查询系统
1. 项目概述:当加密货币数据需要“智能”起来如果你正在开发一个需要实时加密货币数据的应用,或者你是一个数据分析师,每天需要手动从几十个交易所网站和API里抓取价格、市值、交易量,那么你大概率已经对数据源的分散、格式的不统…...