Scissor算法-从含有表型的bulkRNA数据中提取信息进而鉴别单细胞亚群
在做基础实验的时候,研究者都希望能够改变各种条件来进行对比分析,从而探索自己所感兴趣的方向。
在做数据分析的时候也是一样的,我们希望有一个数据集能够附加了很多临床信息/表型,然后二次分析者们就可以进一步挖掘。
然而现实情况总是数据集质量非常不错,但是附加的临床信息/表型却十分有限,这种状况在单细胞数据分析中更加常见。
因此如何将大量的含有临床信息/表型的bulk RNA测序数据和单细胞数据构成联系,这也是算法开发者们所重点关注的方向之一。
其中Scissor算法就可以从含有表型的bulk RNA数据中提取信息去鉴别单细胞亚群。

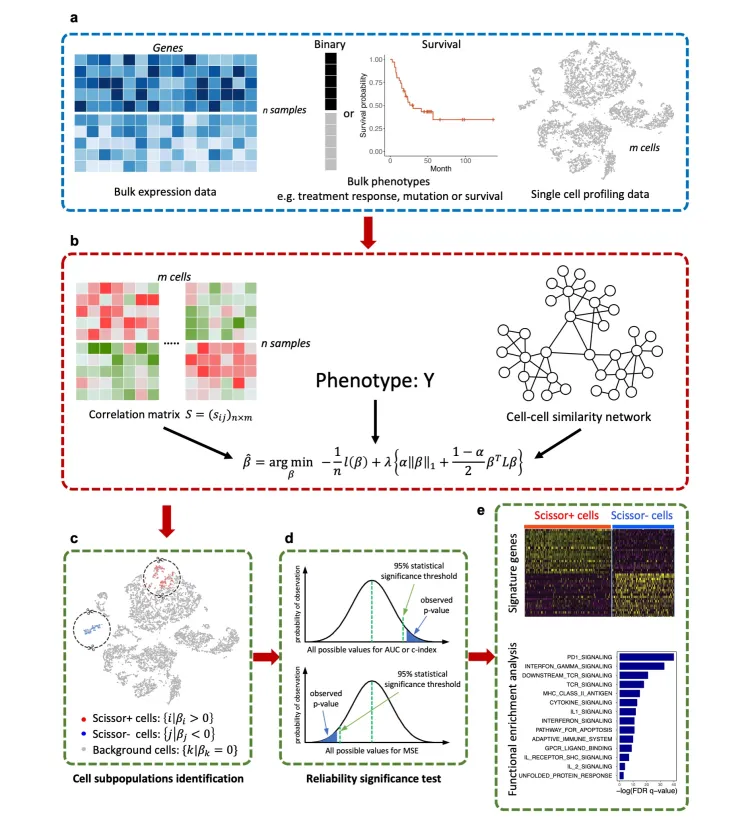
Scissor的分析原理主要是:
基于表达数据计算每个单细胞与bulk样本的相关性,筛选相关性较好的细胞群。
进一步结合表型信息,通过回归模型并加上惩罚项选出最相关的亚群。
原理详情可见:
1、github: https://github.com/sunduanchen/Scissor?tab=readme-ov-file
2、生信技能树教程1:https://mp.weixin.qq.com/s/jC6QTQCfcl_i4tTbFQAq7A
3、生信技能树教程2:https://mp.weixin.qq.com/s/dIYDNDPgIEDUkqqSr56GPg
分析步骤如下:
很多教程展示的是跟生存数据相关的分析,我这里采用二分类数据进行分析。
并且该算法最新一次更新是2021年,如果是使用seruat5版本构建单细胞数据集的话会报错,在进行分析前需要提取Scissor源代码修改一下。
1、导入数据和加载R包
rm(list = ls())
library(Scissor)
library(seurat)
scRNA <- readRDS("scRNA_tumor.rds") #这里采用了自己的处理的单细胞数据
load("step1output.Rdata") #这里也是自己处理的bulkRNA数据
sc_dataset <- scRNA
#dim(sc_dataset)
#[1] 20124 5042
bulk_dataset <- exp
# GSM1310570 GSM1310571 GSM1310572 GSM1310573
#FAM174B 8.232 8.248 7.576 8.708
#AP3S2 5.998 6.079 5.695 6.653
#SV2B 6.107 6.630 5.686 7.886
#RBPMS2 6.718 7.630 7.410 5.762
phenotype <- pd
# title doubling time (days) survival time(months) gender doubling_group OS_group
#GSM1310570 Tumor T_A_001 119 52 female 1 0
#GSM1310571 Tumor T_B_003 98 44 male 0 0
#GSM1310572 Tumor T_C_005 50 3 male 0 0
#GSM1310573 Tumor T_D_007 80 28 male 0 1
#GSM1310574 Tumor T_E_009 197 47 male 1 0
#GSM1310575 Tumor T_F_011 297 52 female 1 0
我这里对OS和double时间进行了分组,变成了二分类数据。后面会单独提取。
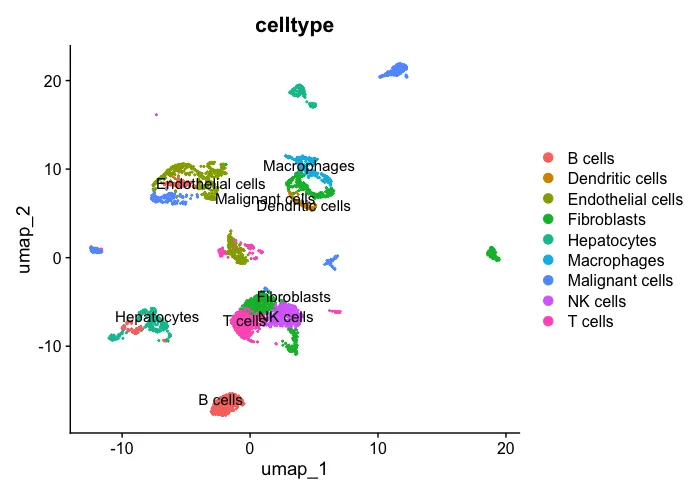
2、先看一下处理好的分群结果
# Check
UMAP_celltype <- DimPlot(sc_dataset, reduction ="umap",group.by="celltype",label = T)
UMAP_celltype
3、运行Scissor,生存数据family设置"cox" ,logistic回归family设置"binomial"。
其中二分类变量在分析前需要设置tag
#提取想要的数据信息
colnames(phenotype)
phenotype <- phenotype[,"doubling_group"]
tag <- c("Quick","Slow")#分析时数据中不能存在na数据,去除或者改成0
#bulk_dataset <- na.omit(bulk_dataset)
bulk_dataset[is.na(bulk_dataset)] <- 0#正式开始分析
infos1 <- Scissor(bulk_dataset, sc_dataset, phenotype, tag = tag,alpha = 0.02, # 默认0.05cutoff = 0.2, #the number of the Scissor selected cells should not exceed 20% of total cells in the single-cell datafamily = "binomial", Save_file = './result.RData')
# Error in as.matrix(sc_dataset@assays$RNA@data) :
# no slot of name "data" for this object of class "Assay5"
Error in as.matrix(sc_dataset@assays$RNA@data) : no slot of name "data" for this object of class "Assay5"
看来这个算法暂不直接适用于seraut5版本,没办法只能提取原代码进行稍作修改,把读取单细胞数据data部分的代码内容增加layer即可,新的代码保存之后再调用。
4、修改之后的代码,命名为RUNscissor
其实就是修改了里边的读取方式:sc_exprs <- as.matrix(sc_dataset@assaysdata)
RUNScissor <- function (bulk_dataset, sc_dataset, phenotype, tag = NULL, alpha = NULL, cutoff = 0.2, family = c("gaussian", "binomial", "cox"), Save_file = "Scissor_inputs.RData", Load_file = NULL)
{library(Seurat)library(Matrix)library(preprocessCore)# 确保 phenotype 是向量phenotype <- as.numeric(phenotype)if (is.null(Load_file)) {common <- intersect(rownames(bulk_dataset), rownames(sc_dataset))if (length(common) == 0) {stop("There is no common genes between the given single-cell and bulk samples.")}if (class(sc_dataset) == "Seurat") {sc_exprs <- as.matrix(sc_dataset@assays$RNA@layers$data)rownames(sc_exprs) <- rownames(sc_dataset)colnames(sc_exprs) <- colnames(sc_dataset)network <- as.matrix(sc_dataset@graphs$RNA_snn)} else {sc_exprs <- as.matrix(sc_dataset)Seurat_tmp <- CreateSeuratObject(sc_dataset)Seurat_tmp <- FindVariableFeatures(Seurat_tmp, selection.method = "vst", verbose = FALSE)Seurat_tmp <- ScaleData(Seurat_tmp, verbose = FALSE)Seurat_tmp <- RunPCA(Seurat_tmp, features = VariableFeatures(Seurat_tmp), verbose = FALSE)Seurat_tmp <- FindNeighbors(Seurat_tmp, dims = 1:10, verbose = FALSE)network <- as.matrix(Seurat_tmp@graphs$RNA_snn)}diag(network) <- 0network[which(network != 0)] <- 1dataset0 <- cbind(bulk_dataset[common, ], sc_exprs[common, ])dataset0 <- as.matrix(dataset0)dataset1 <- normalize.quantiles(dataset0)rownames(dataset1) <- rownames(dataset0)colnames(dataset1) <- colnames(dataset0)Expression_bulk <- dataset1[, 1:ncol(bulk_dataset)]Expression_cell <- dataset1[, (ncol(bulk_dataset) + 1):ncol(dataset1)]X <- cor(Expression_bulk, Expression_cell)quality_check <- quantile(X)print("|**************************************************|")print("Performing quality-check for the correlations")print("The five-number summary of correlations:")print(quality_check)print("|**************************************************|")if (quality_check[3] < 0.01) {warning("The median correlation between the single-cell and bulk samples is relatively low.")}if (family == "binomial") {Y <- as.numeric(phenotype)z <- table(Y)if (length(z) != length(tag)) {stop("The length differs between tags and phenotypes. Please check Scissor inputs and selected regression type.")} else {print(sprintf("Current phenotype contains %d %s and %d %s samples.", z[1], tag[1], z[2], tag[2]))print("Perform logistic regression on the given phenotypes:")}}if (family == "gaussian") {Y <- as.numeric(phenotype)z <- table(Y)if (length(z) != length(tag)) {stop("The length differs between tags and phenotypes. Please check Scissor inputs and selected regression type.")} else {tmp <- paste(z, tag)print(paste0("Current phenotype contains ", paste(tmp[1:(length(z) - 1)], collapse = ", "), ", and ", tmp[length(z)], " samples."))print("Perform linear regression on the given phenotypes:")}}if (family == "cox") {Y <- as.matrix(phenotype)if (ncol(Y) != 2) {stop("The size of survival data is wrong. Please check Scissor inputs and selected regression type.")} else {print("Perform cox regression on the given clinical outcomes:")}}save(X, Y, network, Expression_bulk, Expression_cell, file = Save_file)} else {load(Load_file)}if (is.null(alpha)) {alpha <- c(0.005, 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9)}for (i in 1:length(alpha)) {set.seed(123)fit0 <- APML1(X, Y, family = family, penalty = "Net", alpha = alpha[i], Omega = network, nlambda = 100, nfolds = min(10, nrow(X)))fit1 <- APML1(X, Y, family = family, penalty = "Net", alpha = alpha[i], Omega = network, lambda = fit0$lambda.min)if (family == "binomial") {Coefs <- as.numeric(fit1$Beta[2:(ncol(X) + 1)])} else {Coefs <- as.numeric(fit1$Beta)}Cell1 <- colnames(X)[which(Coefs > 0)]Cell2 <- colnames(X)[which(Coefs < 0)]percentage <- (length(Cell1) + length(Cell2)) / ncol(X)print(sprintf("alpha = %s", alpha[i]))print(sprintf("Scissor identified %d Scissor+ cells and %d Scissor- cells.", length(Cell1), length(Cell2)))print(sprintf("The percentage of selected cell is: %s%%", formatC(percentage * 100, format = "f", digits = 3)))if (percentage < cutoff) {break}cat("\n")}print("|**************************************************|")return(list(para = list(alpha = alpha[i], lambda = fit0$lambda.min, family = family), Coefs = Coefs, Scissor_pos = Cell1, Scissor_neg = Cell2))
}
5、运行RUNScissor
source("~/Desktop/practice/5-Scissor/RUNScissor.R")
infos1 <- RUNScissor(bulk_dataset, sc_dataset, phenotype, tag = tag,alpha = 0.02, # 默认0.05cutoff = 0.2, #the number of the Scissor selected cells should not exceed 20% of total cells in the single-cell datafamily = "binomial", Save_file = './doubling_time.RData')#[1] "|**************************************************|"
#[1] "Performing quality-check for the correlations"
#[1] "The five-number summary of correlations:"
# 0% 25% 50% 75% 100%
#0.03017724 0.29070054 0.32966267 0.37428284 0.70446959
#[1] "|**************************************************|"
#[1] "Current phenotype contains 38 Quick and 43 Slow samples."
#[1] "Perform logistic regression on the given phenotypes:"
#[1] "alpha = 0.02"
#[1] "Scissor identified 202 Scissor+ cells and 690 Scissor- cells."
#[1] "The percentage of selected cell is: 17.691%"
#[1] "|**************************************************|"
Scissor算法首先给出不同比例细胞下单细胞和bulkRNA数据之间的相关性值,如果相关性过低(< 0.01),则会给出warning信息。
此外,表型分组分别为 38例Quick 样本和 43 例Slow样本,数据采用了logistic回归分析,alpha设置为0.02,共获得了202 Scissor+ 细胞和690 Scissor- 细胞。这里的Scissor+ 细胞是指Slow组样本,一般默认表型信息设置为0和1,0代表未发生感兴趣事件,1代表发生了感兴趣事件,在设置tag信息时需要跟表型信息顺序对应起来。
值得重点关注的是这里的alpha和cutoff值。cutoff值则代表所选择细胞的百分比,默认是小于0.2(20%)。Alpha值平衡了 L1范数和网络惩罚的影响,Alpha值越大则惩罚力度也越大从而得到的scissor+/-细胞数也就越少。通常我们应当保证不超过cutoff的范围下,去自定义alpha值。
6、可视化
Scissor_select <- rep(0, ncol(sc_dataset))
names(Scissor_select) <- colnames(sc_dataset)
Scissor_select[infos1$Scissor_pos] <- "Scissor+"
Scissor_select[infos1$Scissor_neg] <- "Scissor-"
sc_dataset <- AddMetaData(sc_dataset, metadata = Scissor_select, col.name = "scissor")
UMAP_scissor <- DimPlot(sc_dataset, reduction = 'umap', group.by = 'scissor',cols = c('grey','royalblue','indianred1'), pt.size = 0.001, order = c("Scissor+","Scissor-"))
UMAP_scissor
table(sc_dataset$scissor)
patchwork::wrap_plots(plots = list(UMAP_celltype,UMAP_scissor), ncol = 2)
saveRDS(sc_dataset,"sc_dataset.rds")
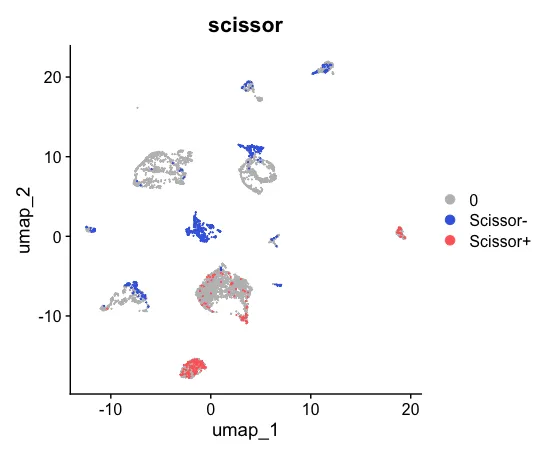
然后可以对两张图片进行对比。
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(希望多多交流)。更多内容可关注公众号:生信方舟
- END -
相关文章:
Scissor算法-从含有表型的bulkRNA数据中提取信息进而鉴别单细胞亚群
在做基础实验的时候,研究者都希望能够改变各种条件来进行对比分析,从而探索自己所感兴趣的方向。 在做数据分析的时候也是一样的,我们希望有一个数据集能够附加了很多临床信息/表型,然后二次分析者们就可以进一步挖掘。 然而现实…...
)
Linux-磁盘空间不足的清理步骤(详细版本)
当 Linux 服务器出现 “no space left on device” 错误时,意味着磁盘空间已满,需要采取一些措施来清理磁盘,可以根据下面步骤依次清理: 1. 检查磁盘使用情况 首先,使用 df 和 du 命令检查磁盘使用情况,找出哪些目录占用了大量空间。 可以通过如下命令来查询 df -h举例…...
go-redis源码解析:连接池原理
1. 执行命令的入口方法 redis也是通过hook执行命令,initHooks时,会将redis的hook放在第一个 通过hook调用到process方法,process方法内部再调用_process 2. 线程池初始化 redis在新建单客户端、sentinel客户端、cluster客户端等,…...

蓝桥杯备赛攻略(怒刷5个月拿省一)
十五届蓝桥杯结束,up也在这次比赛中获得了不错的成绩,为了帮助大家在25年蓝桥杯上获得好的成绩,我将根据今年的经验写一份蓝桥杯的备赛攻略,希望能帮到大家。 参赛准备 蓝桥杯算法赛必须指定一个编程语言赛道报名,也就…...
springboot项目jar包修改数据库配置运行时异常
一、背景 我将软件成功打好jar包了,到部署的时候发现jar包中数据库配置写的有问题,不想再重新打包了,打算直接修改配置文件,结果修改配置后,再通过java -jar运行时就报错了。 二、问题描述 本地项目是springBoot项目…...

倒计时 2 周!CommunityOverCode Asia 2024 IoT Community 专题部分
CommunityOverCode 是 Apache 软件基金会(ASF)的官方全球系列大会,其前身为 ApacheCon。自 1998 年以来,在 ASF 成立之前,ApacheCon 已经吸引了各个层次的参与者,在 300 多个 Apache 项目及其不同的社区中探…...

使用OpenCV在按下Enter键时截图并保存到指定文件夹
使用OpenCV在按下Enter键时截图并保存到指定文件夹 在这篇博客中,我们将介绍如何使用OpenCV库来实现一个简单的功能:在按下Enter键时从摄像头截图并保存到指定的文件夹中。这个功能可以用于各种应用,例如监控系统、视频捕捉等。 前置条件 …...

汇川伺服 (4)FFT、机械特性、闭环、惯量、刚性、抑制振动
一、参数解释 二、FFT 三、机械特性分析 四、多级配方与对象字典 对机组网配方 对象字典 五、InoServoShop 主要是用于调试620P620N将压缩报解压后不需要安装就可以直接使用 六、InoDriveWorkShop 主要是调试660 670 810 520 等系列 惯量识别 Etune Stune 惯量比调试 大惯…...

Unity3D中使用并行的Job完成筛选类任务详解
在Unity3D开发中,处理大量数据或执行复杂计算时,性能往往成为制约因素。为了提升游戏或应用的性能,Unity提供了强大的Job System,它允许开发者利用多线程和并行计算来优化数据处理过程。本文将详细介绍如何在Unity中使用并行的Job…...

汽车信息安全--欧盟汽车法规
目录 General regulation 信息安全法规 R155《网络安全及网络安全管理系统》解析 R156《软件升级与软件升级管理系统》解析 General regulation 欧洲的汽车行业受到一系列法律法规的约束,包括 各个方面包括: 1.安全要求:《通用安全条例&a…...

@SpringBootApplication 注解
什么是 SpringBootApplication SpringBootApplication 是 Spring Boot 提供的一个核心注解,它是一个组合注解,用于简化 Spring Boot 应用程序的配置。这个注解通常标注在主类上,用于标识一个 Spring Boot 应用的入口。通过这个注解ÿ…...

java项目总结4
目录 1.正则表达式 2.爬虫 3.时间 4.包装类 5.工具类之Arrays 6.Lambda 1.正则表达式 用于验证字符串是否满足自己所需要的规则。方法:matches 注意:\在Java中有特殊涵义,是将其它的意思本来化,假设"是用来引…...

JavaScript中的数组方法总结+详解
##JavaScript中的数组方法总结详解 用来总结和学习,便于自己查找 文章目录 一、数组是什么? 二、改变原数组的api方法? 2.1 push() 在末端添加 2.2 pop࿰…...

环境变量Path
PATH 是一个环境变量,它在操作系统中扮演着非常重要的角色。它定义了系统在查找可执行文件时应该搜索的目录列表。当你在命令行中输入一个命令时,操作系统会按照 PATH 变量中定义的目录顺序来查找这个命令对应的可执行文件。 主要作用 查找可执行文件&a…...

基于jeecgboot-vue3的Flowable流程-集成仿钉钉流程(四)支持json和xml的显示
因为这个项目license问题无法开源,更多技术支持与服务请加入我的知识星球。 1、相应的界面前端代码 <template><div class"formDesign"><FlowDesign :process"process" :fields"fields" :readOnly"readOnly&quo…...

【k8s安装redis】k8s安装单机版redis实现高性能高可用
文章目录 简介一.条件及环境说明:二.需求说明:三.实现原理及说明四.详细步骤4.1.创建configmap 配置文件4.2.创建StatefulSet 配置4.3.创建service headless 配置 五.安装说明 简介 本文将根据在k8s环境中搭建【伪】单机模式的redis实例。由于共享存储的…...

Scala 数据类型
Scala 数据类型 Scala 是一种多范式的编程语言,它结合了面向对象和函数式编程的特点。在 Scala 中,数据类型是构建复杂程序的基础。本文将详细介绍 Scala 中的数据类型,包括其分类、特点以及使用方法。 数据类型分类 Scala 中的数据类型可…...

Java Executors类的9种创建线程池的方法及应用场景分析
在Java中,Executors 类提供了多种静态工厂方法来创建不同类型的线程池。在学习线程池的过程中,一定避不开Executors类,掌握这个类的使用、原理、使用场景,对于实际项目开发时,运用自如,以下是一些常用的方法…...

LY/T 3359-2023 耐化学腐蚀高压装饰层积板检测
耐化学腐蚀高压装饰层积板是指用酚醛树脂浸渍的层状植物纤维材料为基材,与涂布以丙烯酸树脂为主体的装饰纸的饰面层,在高温高压下层积压制而成的具有化学腐蚀功能的高压装饰层积板。 LY/T 3359-2023 耐化学腐蚀高压装饰层积板检测项目: 测试…...

【linux/shell】如何创建脚本函数库并在其他脚本中调用
目录 1. 创建脚本库文件 2. 修改脚本库权限,使脚本库可执行 3. 在其他脚本中调用脚本库 4. 使用环境变量或.bashrc 5. 使用Shellcheck 6. 编写注释及说明文档 在Shell中创建和使用脚本库通常涉及以下几个步骤: 1. 创建脚本库文件 脚本库通常是包…...

Python开发者必备:Awesome清单高效选型与实战指南
1. 项目概述:一份Python开发者的“藏宝图”如果你是一名Python开发者,无论是刚入门的新手,还是摸爬滚打多年的老手,我相信你都曾有过这样的时刻:面对一个具体的开发需求,比如想找一个好用的Web框架、一个高…...

魔兽争霸3终极优化工具:5分钟搞定所有兼容性问题
魔兽争霸3终极优化工具:5分钟搞定所有兼容性问题 【免费下载链接】WarcraftHelper Warcraft III Helper , support 1.20e, 1.24e, 1.26a, 1.27a, 1.27b 项目地址: https://gitcode.com/gh_mirrors/wa/WarcraftHelper 还在为《魔兽争霸3》在现代电脑上的各种问…...

从SELinux到AppArmor:聊聊Linux内核安全模块LSM的实战选择与避坑指南
从SELinux到AppArmor:Linux内核安全模块实战选择与避坑指南 在当今云计算和容器化技术蓬勃发展的背景下,Linux系统的安全性变得前所未有的重要。作为系统管理员或DevOps工程师,我们常常需要在安全性和易用性之间寻找平衡点。Linux内核安全模块…...

实测Taotoken多模型API的响应延迟与稳定性观感
🚀 告别海外账号与网络限制!稳定直连全球优质大模型,限时半价接入中。 👉 点击领取海量免费额度 实测Taotoken多模型API的响应延迟与稳定性观感 作为一名需要频繁调用大模型API的开发者,选择一个稳定、可靠的接入平台…...
)
SITS 2026议程背后隐藏的3条技术演进红线(附Gartner/IEEE双认证时间轴对比图)
更多请点击: https://intelliparadigm.com 第一章:2026奇点智能技术大会完整议程曝光:SITS 2026四大看点抢先看 全球瞩目的奇点智能技术大会(Singularity Intelligence Technology Summit, SITS)将于2026年5月12–15日…...

LayerDivider:用AI智能算法重新定义图像分层技术
LayerDivider:用AI智能算法重新定义图像分层技术 【免费下载链接】layerdivider A tool to divide a single illustration into a layered structure. 项目地址: https://gitcode.com/gh_mirrors/la/layerdivider 在数字设计领域,图像分层是创意工…...

QueryExcel:批量Excel数据检索的自动化解决方案
QueryExcel:批量Excel数据检索的自动化解决方案 【免费下载链接】QueryExcel 多Excel文件内容查询工具。 项目地址: https://gitcode.com/gh_mirrors/qu/QueryExcel 在数据驱动的现代办公环境中,Excel文件已成为信息存储的主要载体。然而…...

taotoken官方折扣活动下tokenplan套餐的性价比分析
🚀 告别海外账号与网络限制!稳定直连全球优质大模型,限时半价接入中。 👉 点击领取海量免费额度 taotoken官方折扣活动下tokenplan套餐的性价比分析 效果展示类,结合平台近期的官方折扣活动,客观分析选择不…...

如何永久保存微信聊天记录?WeChatMsg完整指南带你一键备份
如何永久保存微信聊天记录?WeChatMsg完整指南带你一键备份 【免费下载链接】WeChatMsg 提取微信聊天记录,将其导出成HTML、Word、CSV文档永久保存,对聊天记录进行分析生成年度聊天报告 项目地址: https://gitcode.com/GitHub_Trending/we/W…...

Windows与Office一键激活终极指南:KMS_VL_ALL_AIO智能脚本完整教程
Windows与Office一键激活终极指南:KMS_VL_ALL_AIO智能脚本完整教程 【免费下载链接】KMS_VL_ALL_AIO Smart Activation Script 项目地址: https://gitcode.com/gh_mirrors/km/KMS_VL_ALL_AIO 还在为Windows系统激活和Office办公软件激活而烦恼吗?…...