数据分析:微生物数据的荟萃分析框架
介绍
Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer提供了一种荟萃分析的框架,它主要基于常用的Wilcoxon rank-sum test和Blocked Wilcoxon rank-sum test 方法计算显著性,再使用分位数计算分组间的倍数变化,最后通过AUC判断物种的区分分组的能力。最后通过热图和森林图展示筛选到的在不同研究和荟萃分析均有差异的物种。
该框架可用于同类型的微生物荟萃分析。
加载R包
#| warning: false
#| message: falselibrary(tidyverse)
library(readr)
library(coin)
library(pROC)
library(RColorBrewer)
library(cowplot)# rm(list = ls())
options(stringsAsFactors = F)
options(future.globals.maxSize = 1000 * 1024^2)
导入数据
数据下载百度云盘链接: https://pan.baidu.com/s/1VS6S8p5s20vwZ6FyILYoaQ
提取码: g4y3
-
物种表达谱数据
-
样本分组信息
#| warning: false
#| message: falsefeat.all <- read.table("./data/meta-CRC-2019/feat_rel_crc.tsv", sep='\t', header=TRUE, stringsAsFactors = FALSE, check.names = FALSE, quote='') %>%as.matrix()meta <- read_tsv('./data/meta-CRC-2019/meta_crc.tsv', show_col_types = FALSE)
- 其他参数(过滤和检验结果)
#| warning: false
#| message: falsealpha.meta <- 1e-05
alpha.single.study <- 0.005
mult.corr <- 'fdr'
pr.cutoff <- 0.05
log.n0 <- 1e-05
log.n0.func <- 1e-08
study.cols <- c('#2FBFBF', '#177254', '#F2CC30', '#74B347', '#8265CC')
数据预处理
-
提出研究名称
studies -
设置
block分组,用于后续检验
#| warning: false
#| message: falsestudies <- meta %>% dplyr::pull(Study) %>% unique# block for colonoscopy and study as well
meta <- meta %>%dplyr::filter(!is.na(Sampling_rel_to_colonoscopy)) %>%dplyr::mutate(block = ifelse(Study != 'CN-CRC', Study, paste0(Study, '_', Sampling_rel_to_colonoscopy)))feat.all <- feat.all[, meta$Sample_ID]
荟萃分析
荟萃分析采用了Wilcoxon rank-sum test和Blocked Wilcoxon rank-sum test 两种方法对单个研究和合并所有研究做显著性检验。本次需要计Foldchange(FC),单个研究的pvalue + 所有研究的pvalue(p.val)和单个研究和所有研究的AUC(aucs),以下是该代码的计算过程:
-
先使用Wilcoxon rank-sum test计算每个研究的每个物种在case/control之间的显著性检验结果;
-
再通过roc函数计算每个研究的每个物种在case/control之间的判别效果;
-
接着通过分位数quantile计算每个研究的每个物种在case/control之间的倍数变化;
-
然后通过Blocked Wilcoxon rank-sum test计算所有研究的荟萃差异检验结果;
-
最后计算所有研究的平均倍数变化作为整体倍数变化和通过roc函数计算每个物种在case/control之间的判别效果。
#| warning: false
#| message: falsep.val <- matrix(NA, nrow = nrow(feat.all), ncol = length(studies)+1, dimnames = list(row.names(feat.all), c(studies, 'all')))
fc <- p.val
aucs.mat <- p.val
aucs.all <- vector('list', nrow(feat.all))cat("Calculating effect size for every feature...\n")
pb <- txtProgressBar(max = nrow(feat.all), style = 3)# caluclate wilcoxon test and effect size for each feature and study
for (f in row.names(feat.all)) {# for each studyfor (s in studies) {x <- feat.all[f, meta %>% dplyr::filter(Study == s) %>% dplyr::filter(Group=='CRC') %>% dplyr::pull(Sample_ID)]y <- feat.all[f, meta %>% dplyr::filter(Study==s) %>% dplyr::filter(Group=='CTR') %>% dplyr::pull(Sample_ID)]# Wilcoxon: 对单个研究的单个物种检验p.val[f, s] <- wilcox.test(x, y, exact=FALSE)$p.value# AUC:评估每个物种区分分组的能力aucs.all[[f]][[s]] <- c(roc(controls=y, cases=x, direction='<', ci=TRUE, auc=TRUE)$ci)aucs.mat[f, s] <- c(roc(controls=y, cases=x, direction='<', ci=TRUE, auc=TRUE)$ci)[2]# FC:使用10分位数计算每个物种的相对丰度再计算Foldchange结果q.p <- quantile(log10(x+log.n0), probs = seq(.1, .9, .05))q.n <- quantile(log10(y+log.n0), probs = seq(.1, .9, .05))fc[f, s] <- sum(q.p - q.n)/length(q.p)}# calculate effect size for all studies combined# Wilcoxon + blocking factor:计算所有研究混合在一起的检验结果d <- data.frame(y = feat.all[f,], x = meta$Group, block = meta$block) %>%dplyr::mutate(x = factor(x),block = factor(block))p.val[f, 'all'] <- coin::pvalue(wilcox_test(y ~ x | block, data = d))# other metricsx <- feat.all[f, meta %>% dplyr::filter(Group=='CRC') %>% dplyr::pull(Sample_ID)]y <- feat.all[f, meta %>% dplyr::filter(Group=='CTR') %>% dplyr::pull(Sample_ID)]# FC: 取所有样本的平均FC结果fc[f, 'all'] <- mean(fc[f, studies])# AUC:合并数据集每个物种区分不同分组样本的能力aucs.mat[f, 'all'] <- c(roc(controls=y, cases=x, direction='<', ci=TRUE, auc=TRUE)$ci)[2]# progressbarsetTxtProgressBar(pb, (pb$getVal()+1))
}
cat('\n')# multiple hypothesis correction
p.adj <- data.frame(apply(p.val, MARGIN=2, FUN=p.adjust, method=mult.corr),check.names = FALSE)
查看结果
查看上述荟萃分析的结果
#| warning: false
#| message: falsehead(p.adj)head(aucs.mat)head(fc)

画图
文章给出的图分成两部分,上部分是热图形式,下半部是森林图。
- 热图: 展示不同研究显著差异的物种
#| warning: false
#| message: falsespecies.heatmap <- rownames(p.adj)[which(p.adj$all < alpha.single.study)]fc.sign <- sign(fc)
fc.sign[fc.sign == 0] <- 1p.val.signed <- -log10(p.adj[species.heatmap,"all", drop=FALSE]) * fc.sign[species.heatmap, 'all']top.markers <- rownames(p.val.signed[is.infinite(p.val.signed$all) , , drop=FALSE])
p.val.signed[top.markers, 'all'] <- 100 + aucs.mat[top.markers, 'all']species.heatmap.orderd <- rownames(p.val.signed[order(p.val.signed$all), , drop=FALSE])# take only those
fc.mat.plot <- fc[species.heatmap.orderd, ] %>% as.data.frame()
p.vals.plot <- p.adj[species.heatmap.orderd, ]# ##############################################################################
# prepare plotting# colorscheme for fc heatmap
mx <- max(abs(range(fc.mat.plot, na.rm=TRUE)))
mx <- ifelse(round(mx, digits = 1) < mx, round(mx, digits = 1) + 0.1, round(mx, digits = 1))
brs <- seq(-mx, mx, by=0.05)
num.col.steps <- length(brs) - 1
n <- floor(0.45*num.col.steps)
col.hm <- c(rev(colorRampPalette(brewer.pal(9, 'Blues'))(n)),rep('#FFFFFF', num.col.steps-2*n),colorRampPalette(brewer.pal(9, 'Reds'))(n))
# color scheme for pval heatmap
alpha.breaks <- c(1e-06, 1e-05, 1e-04, 1e-03, 1e-02, 1e-01)
p.vals.bin <- data.frame(apply(p.vals.plot, 2, FUN=.bincode, breaks = c(0, alpha.breaks, 1), include.lowest = TRUE),check.names = FALSE)
p.val.greys <- c(paste0('grey', round(seq(from=10, to=80, length.out = length(alpha.breaks)))), 'white')
names(p.val.greys) <- as.character(1:7)# function to plot both into a grid
plot.single.study.heatmap <- function(x) {# x = "FR-CRC"df.plot <- tibble(species = factor(rownames(p.vals.plot), levels = rev(rownames(p.vals.plot))),p.vals = as.factor(p.vals.bin[[x]]),fc = fc.mat.plot[[x]])g1 <- df.plot %>% ggplot(aes(x = species, y = 1, fill = fc)) + geom_tile() + theme_minimal() + theme(axis.text = element_blank(),axis.ticks = element_blank(),axis.title = element_blank(), panel.grid = element_blank(),panel.background = element_rect(fill=NULL, colour='black'),plot.margin = unit(c(0, 0, 0, 0), 'cm')) + scale_y_continuous(expand = c(0, 0)) + scale_fill_gradientn(colours=col.hm, limits=c(-mx, mx), guide=FALSE)g2 <- df.plot %>% ggplot(aes(x=species, y=1, fill=p.vals)) +geom_tile() + theme_minimal() + theme(axis.text = element_blank(),axis.ticks = element_blank(),axis.title = element_blank(), panel.grid = element_blank(),panel.background = element_rect(fill=NULL, colour='black'),plot.margin = unit(c(0, 0, 0, 0), 'cm')) + scale_y_continuous(expand = c(0, 0)) + scale_fill_manual(values=p.val.greys, na.value='white', guide=FALSE)g.return <- plot_grid(g2, g1, ncol = 1, rel_heights = c(0.25, 0.75))return(g.return)
}# ##############################################################################
# plot# p.value histogram
g1 <- tibble(species = factor(rownames(p.vals.plot), levels = rev(rownames(p.vals.plot))),p.vals = -log10(p.vals.plot$all),colour = p.vals > 5) %>% ggplot(aes(x = species, y = p.vals, fill = colour)) + geom_bar(stat = 'identity') + theme_classic() + xlab('Gut microbial species') + ylab('-log10(q-value)') + theme(panel.grid.major = element_blank(),panel.grid.minor = element_blank(),axis.ticks.x = element_blank(),axis.text.x = element_blank(),panel.background = element_rect(fill = NULL, color = 'black')) + scale_y_continuous(limits = c(0, 15), expand = c(0, 0)) + scale_x_discrete(position = 'top') + scale_fill_manual(values = c('lightgrey', 'darkgrey'), guide = FALSE)g.lst <- lapply(studies, plot.single.study.heatmap)pl1 <- plot_grid(g1, g.lst[[1]], g.lst[[2]], g.lst[[3]], g.lst[[4]],g.lst[[5]], ncol = 1, align = 'v', rel_heights = c(0.3, rep(0.12, 5)))pl1

结果:差异物种在不同研究和整合数据的倍数变化和显著性结果
-
最上图是物种在整合数据的显著性结果(adjustedPvalue);
-
接下来的热图是物种在单个研究的显著性结果(上半部)和倍数变化(下半部:红色是富集在CRC,蓝色是CTRL);
-
森林图: 不同物种在每个研究区分case/control的能力 (通过alpha.meta更严格筛选)
#| warning: false
#| message: false# select and order
marker.set <- rownames(p.val.signed)[abs(p.val.signed$all) > -log10(alpha.meta)]
p.val.signed.red <- p.val.signed[marker.set, ,drop=FALSE]
marker.set.orderd <- rev(rownames(p.val.signed.red[order(p.val.signed.red$all),,drop=FALSE]))# extract those from the auc list
df.plot <- tibble()
for (i in marker.set.orderd){for (s in studies){temp <- aucs.all[[i]][[s]]df.plot <- bind_rows(df.plot, tibble(species=i, study=s,low=temp[1], auc=temp[2], high=temp[3]))}
}df.plot <- df.plot %>% dplyr::mutate(species = factor(species, levels = marker.set.orderd)) %>% dplyr::mutate(study = factor(study, levels = studies))# plot everything
pl2 <- df.plot %>% ggplot(aes(x = study, y = auc)) + geom_linerange(aes(ymin = low, ymax = high), color = 'lightgrey') + geom_point(pch = 23, aes(fill = study)) + facet_grid(~species, scales = 'free_x', space = 'free') + theme_minimal() + scale_y_continuous(limits=c(0, 1)) + theme(panel.grid.major.x = element_blank(),axis.ticks.x = element_blank(),axis.text.x = element_blank(),strip.text = element_text(angle=90, hjust=0)) + scale_fill_manual(values = study.cols, guide = FALSE) + ylab('AUROC') + xlab('Gut microbial species')pl2

- 合并图: 最后文章呈现的图是经过修改的
#| warning: false
#| message: falsecowplot::plot_grid(pl1, pl2, ncol = 1)
总结
在进行荟萃分析时,本研究采用了一种特定的统计方法——Blocked Wilcoxon rank-sum test,以评估和整合不同研究中的case/control物种的显著性结果。该方法特别适用于处理微生物数据这类稀疏性数据集,因为它能够在计算两组之间的倍数变化时有效避免零值过多的问题。通过使用分位数方法,研究者能够更准确地估计和比较不同组之间的差异,从而提高了分析结果的可靠性和有效性。
对于类似类型的研究,研究者可以采用与本研究相似的分析框架进行荟萃分析。这包括以下几个关键步骤:
- 数据的收集与整理:确保收集到的数据是高质量的,并且适合进行荟萃分析。
- 选择合适的统计方法:根据数据的特点选择合适的统计检验方法,如Blocked Wilcoxon rank-sum test,以确保分析的准确性。
- 数据处理:对于稀疏数据,采用分位数方法来处理零值过多的问题,以提高分析的稳健性。
- 结果的整合与解释:将不同研究的结果进行整合,并采用适当的统计方法来评估整体的显著性。
通过遵循这样的框架,研究者可以对类似主题的研究进行系统性地分析和比较,从而为该领域的研究提供更深入的见解。
相关文章:
数据分析:微生物数据的荟萃分析框架
介绍 Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer提供了一种荟萃分析的框架,它主要基于常用的Wilcoxon rank-sum test和Blocked Wilcoxon rank-sum test 方法计算显著性,再使用分…...
Django—admin后台管理
Django官网 https://www.djangoproject.com/ 如果已经有了Django跳过这步 安装Django: 如果你还没有安装Django,可以通过Python的包管理器pip来安装: pip install django 创建项目: 使用Django创建一个新的项目: …...
数字图像处理中的常用特殊矩阵及MATLAB应用
一、前言 Matlab的名称来源于“矩阵实验室(Matrix Laboratory)”,其对矩阵的操作具有先天性的优势(特别是相对于C语言的数组来说)。在数字图像处理中,为了提高编程效率,我们可以使用多种方式来创…...
精彩案例剖析一)
vue侦听器(Watch)精彩案例剖析一
目录 watch介绍 监视普通数据类型 监视对象类型 watch介绍 在 Vue 中,watch主要用于监视数据的变化,并执行相应操作。一旦被监视的属性发生变化,回调函数将自动被触发。当在 Vue 中使用watch来响应数据变化时,首先要清楚,watch本质上是一个对象,且必须以对象的…...

HTTP 协议浅析
HTTP(HyperText Transfer Protocol,超文本传输协议)是应用层最重要的协议之一。它定义了客户端和服务器之间的数据传输方式,并成为万维网(World Wide Web)的基石。本文将深入解析 HTTP 协议的基础知识、工作…...
VsCode | 让空文件夹始终展开不折叠
文章目录 1 问题引入2 解决办法3 效果展示 1 问题引入 可能很多小伙伴更新VsCode或者下载新版本时候 ,创建的文件 会出现xxx文件夹/xxx文件夹,看着很不舒服,所以该如何展开所有空文件夹呢? 2 解决办法 找到VsCode的设置 &…...
Centos7_Minimal安装Cannot find a valid baseurl for repo: base/7/x86_6
问题 运行yum报此问题 就是没网 解决方法 修改网络信息配置文件,打开配置文件,输入命令: vi /etc/sysconfig/network-scripts/ifcfg-网卡名字把ONBOOTno,改为ONBOOTyes 重启网卡 /etc/init.d/network restart 网路通了...
Spark_Oracle_II_Spark高效处理Oracle时间数据:通过JDBC桥接大数据与数据库的分析之旅
接前文背景, 当需要从关系型数据库(如Oracle)中读取数据时,Spark提供了JDBC连接功能,允许我们轻松地将数据从Oracle等数据库导入到Spark DataFrame中。然而,在处理时间字段时,可能会遇到一些挑战…...
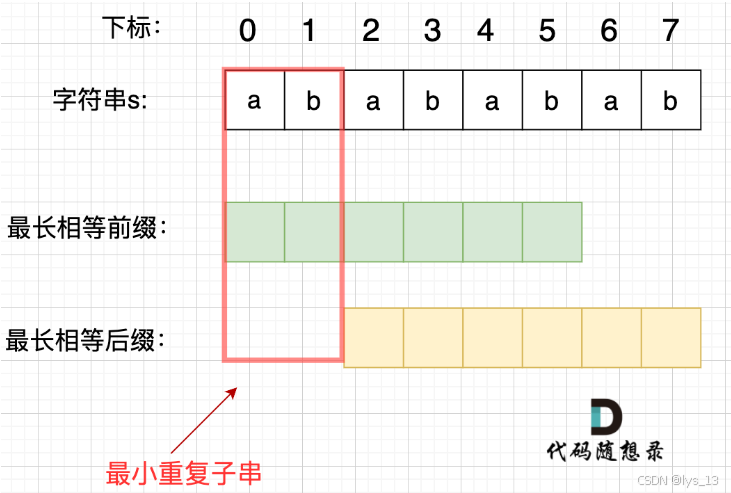
力扣 459重复的子字符串
思路: KMP算法的核心是求next数组 next数组代表的是当前字符串最大前后缀的长度 而求重复的子字符串就是求字符串的最大前缀与最大后缀之间的子字符串 如果这个子字符串是字符串长度的约数,则true /** lc appleetcode.cn id459 langcpp** [459] 重复…...

MyBatis XML配置文件
目录 一、引入依赖 二、配置数据库的连接信息 三、实现持久层代码 3.1 添加mapper接口 3.2 添加UserInfoXMLMapper.xml 3.3 增删改查操作 3.3.1 增(insert) 3.3.2 删(delete) 3.3.3 改(update) 3.3.4 查(select) 本篇内容仍然衔接上篇内容,使用的代码及案…...

读写RDS或RData等不同格式的文件,包括CSV和TXT、Excel的常见文件格式,和SPSS、SAS、Stata、Minitab等统计软件的数据文件
R语言是数据分析和科学计算的强大工具,其丰富的函数和包使得处理各种数据格式变得相对简单。在本文中,我们将详细介绍如何使用R语言的函数命令读取和写入不同格式的文件,包括RDS或RData格式文件、常见的文本文件(如CSV和TXT)、Excel文件,和和SPSS、SAS、Stata、Minitab等…...
视频支持格式)
Android 支持的媒体格式,(二)视频支持格式
视频支持格式: 格式编码器解码器具体说明文件类型 容器格式H.263是是对 H.263 的支持在 Android 7.0 及更高版本中并非必需• 3GPP (.3gp) • MPEG-4 (.mp4) • Matroska (.mkv)H.264 AVC Baseline Profile (BP)Android 3.0 及以上版本是 • 3GPP (.3gp) • MPEG-4…...

密码学原理精解【8】
文章目录 概率分布哈夫曼编码实现julia官方文档建议的变量命名规范:julia源码 熵一、信息熵的定义二、信息量的概念三、信息熵的计算步骤四、信息熵的性质五、应用举例 哈夫曼编码(Huffman Coding)基本原理编码过程特点应用具体过程1. 排序概…...
2024年钉钉杯大数据竞赛A题超详细解题思路+python代码手把手保姆级运行讲解视频+问题一代码分享
初赛A:烟草营销案例数据分析 AB题综合难度不大,难度可以视作0.4个国赛,题量可以看作0.35个国赛题量。适合于国赛前队伍练手,队伍内磨合。竞赛获奖率50%,八月底出成绩,参赛人数3000队左右。本文将为大家进行…...
unity2D游戏开发01项目搭建
1新建项目 选择2d模板,设置项目名称和存储位置 在Hierarchy面板右击,create Empty 添加组件 在Project视图中右键新建文件夹 将图片资源拖进来(图片资源在我的下载里面) 点击Player 修改属性,修好如下 点击Sprite Editor 选择第二…...

删除的视频怎样才能恢复?详尽指南
在日常生活中,我们有时会不小心删除一些重要的视频文件,或者在整理存储空间时不慎丢失了珍贵的记忆片段。这时候,我们可以通过一些数据恢复工具和技巧,找回这些被删除的视频。本文将详细介绍几种常见且有效的视频恢复方法…...

LeetCode160 相交链表
前言 题目: 160. 相交链表 文档: 代码随想录——链表相交 编程语言: C 解题状态: 没思路… 思路 依旧是双指针法,很巧妙的方法,有点想不出来。 代码 先将两个链表末端对齐,然后两个指针齐头并…...

高性能响应式UI部件DevExtreme v24.1.4全新发布
DevExtreme拥有高性能的HTML5 / JavaScript小部件集合,使您可以利用现代Web开发堆栈(包括React,Angular,ASP.NET Core,jQuery,Knockout等)构建交互式的Web应用程序。从Angular和Reac,…...

Python实现Java mybatis-plus 产生的SQL自动化测试SQL速度和判断SQL是否走索引
Python实现Java mybatis-plus 产生的SQL自动化测试SQL速度和判断SQL是否走索引 文件目录如下 │ sql_speed_test.py │ ├─input │ data-report_in_visit_20240704.log │ resource_in_sso_20240704.log │ └─outputdata-report_in_visit_20240704.cs…...
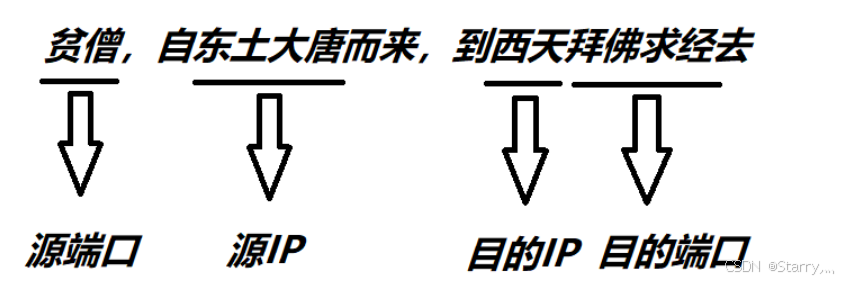
UDP的报文结构及其注意事项
1. 概述 UDP(User Datagram Protocol)是一种无连接的传输层协议,它提供了一种简单的数据传输服务,不保证数据的可靠传输。在网络通信中,UDP通常用于一些对实时性要求较高、数据量较小、传输延迟较低的应用,…...

Miniblink49:如何在5分钟内将浏览器内核嵌入你的C++应用?
Miniblink49:如何在5分钟内将浏览器内核嵌入你的C应用? 【免费下载链接】miniblink49 a lighter, faster browser kernel of blink to integrate HTML UI in your app. 一个小巧、轻量的浏览器内核,用来取代wke和libcef 项目地址: https://…...

工程定制丙级管道井门 物业机房通用款式
工程定制丙级管道井门,作为高层住宅、商业楼宇、物业机房强弱电井的专用消防配套设施,严格遵循国标消防规范生产,是建筑管井防火分隔、安全防护的核心产品。这款丙级管道井门采用钢制一体成型工艺,结构扎实不易变形,具…...

HsMod终极指南:50+功能全面解锁炉石传说模改插件
HsMod终极指南:50功能全面解锁炉石传说模改插件 【免费下载链接】HsMod Hearthstone Modification Based on BepInEx 项目地址: https://gitcode.com/GitHub_Trending/hs/HsMod HsMod是一款基于BepInEx框架开发的炉石传说模改插件,通过50多项实用…...

5分钟搞定安卓APK签名:SignatureTools图形化签名工具终极指南
5分钟搞定安卓APK签名:SignatureTools图形化签名工具终极指南 【免费下载链接】SignatureTools 🎡使用JavaFx编写的安卓Apk签名&渠道写入工具,方便快速进行v1&v2签名。 项目地址: https://gitcode.com/gh_mirrors/si/SignatureTool…...

ROS小车转弯卡顿?手把手教你用Python搞定cmd_vel到阿克曼模型的平滑转换
ROS小车转弯卡顿?Python实现cmd_vel到阿克曼模型的平滑转换实战 当你在Gazebo仿真或实际运行ROS控制的阿克曼转向小车时,是否遇到过车体转弯时"一耸一耸"、运动不连贯的尴尬情况?这种卡顿现象往往源于cmd_vel指令到阿克曼运动模型转…...

抖音视频批量下载难题如何解决?douyin-downloader开源工具完整指南
抖音视频批量下载难题如何解决?douyin-downloader开源工具完整指南 【免费下载链接】douyin-downloader A practical Douyin downloader for both single-item and profile batch downloads, with progress display, retries, SQLite deduplication, and browser fa…...

如何利用Dask集成ydata-profiling实现大规模数据处理:2024终极指南
如何利用Dask集成ydata-profiling实现大规模数据处理:2024终极指南 【免费下载链接】fg-data-profiling 1 Line of code data quality profiling & exploratory data analysis for Pandas and Spark DataFrames. 项目地址: https://gitcode.com/gh_mirrors/y…...

Claude Code 工具提示词全拆解:AI Agent、Prompt Engineering、工具调用、上下文工程、自动化编程的底层逻辑
开篇导读很多人做 AI Agent 时,最容易盯着模型参数、系统提示词、工具数量,却忽略了一个非常关键的细节:每一个工具自己的提示词。它看起来只是一个 description 字段,实际上却在悄悄决定模型什么时候用工具、怎样用工具、不能做什…...

28纳米工艺下SAR ADC架构的核心优势与设计要点
1. SAR ADC架构在28纳米工艺中的核心优势在移动SoC设计中,模数转换器(ADC)的性能直接影响着整个系统的功耗和面积效率。随着工艺节点演进至28纳米及以下,逐次逼近型(SAR)ADC架构展现出三大核心优势…...
)
YOLO26驱动的足球比赛多目标检测系统:球员、守门员、裁判与足球的实时识别(项目源码+数据集+模型权重+UI界面+python+深度学习+远程环境部署)
摘要 足球作为全球最受欢迎的体育运动之一,其数字化分析对于战术研究、运动员评估和比赛裁判具有重要意义。本文基于YOLO目标检测算法,构建了一套足球运动员识别检测系统,实现对比赛场景中足球、守门员、球员和裁判四类目标的自动检测与定位…...