NCR+可变电荷块3——NCB/cell绘图1
文献method参考:

蛋白质序列数据从uniprot中获取

https://www.uniprot.org/uniprotkb/P46013/entry

https://www.uniprot.org/uniprotkb/P06748/entry、
1,电荷分布计算:
Charge distribution was calculated as the sum of the charges (Arg and Lys, +1; Glu and Asp, −1; phospho-Ser and phospho-Thr, −2) in the indicated window range. A charge block was designated when the area of the charge plot (window size: 35 amino acids) was larger than 20.
(1)电荷分布是计算指定窗口window内净电荷之和,所以dict对应关系是1个window对应1个电荷总和,所以电荷分布图实际上就是绘制(x,y)=(窗口编号,该窗口净电荷总和)的折线图
①首先计算给定窗口内每个氨基酸的净电荷是简单的:
碱性带正电aa:KR,但是没有组氨酸H?
酸性带负电aa:DE
至于磷酸化的氨基酸没有数据可以验证(我们只有序列数据,没法获取,需要实际in vivo数据,也许可以从蛋白质晶体数据库上获取?)
可以另外开一个磷酸化的字典dict,存放不同的aa字符与带电荷的对应关系,其实这样的PTM的字典可以开很多
**②问题是计算电荷分布的时候窗口开多大?
**是method里指定的win=35吗?
这里很容易被误导,实际上不是35。
35指的是charge plot已经绘制出来之后,然后计算特定区域内charge plot的area面积,实际上应该需要进行积分。
有个问题,如何理解这里的area>20,首先要理解block,肯定是单一电荷性质的,所以肯定是看的积分之后的净area,比如说是计算对应的正电荷的正面积之后,以及负电荷的负面积,然后计算这个窗口的电荷总和,所以实际上就是电荷面积计算的一个净net值的绝对值。
**所以在计算charge plot中的window窗口应该设置为多大?
**













可以看到实际上每一幅图指定的window窗口大小尺寸都是25


但是在提供的附近材料(审稿意见答复)中又说是35
当然仅仅只是获取charge plot的话窗口定义多大多小都不影响,因为只是展示:
看复现效果:Ki-67


16个RD重复单元,每个大概在110个aa左右
我们只看R12

实际上可以看到对应紫色那块,也就是K167R 12


实际上大小是112
示例代码如下:
def calculate_charge_distribution(sequence, phospho_sites, window_size=35):"""计算蛋白质电荷分布Args:sequence: 蛋白质氨基酸序列 (字符串)phospho_sites: 磷酸化位点列表 (例如:[10, 25, 50]),位点编号从1开始,蛋白质序列index是1-indexwindow_size: 滑动窗口大小 (整数)Returns:一个列表,包含每个窗口的净电荷。"""charges = {'R': 1, 'K': 1, 'E': -1, 'D': -1, 'S': 0, 'T': 0} #因为原文考虑到了丝氨酸以及苏氨酸的磷酸化与否对蛋白质电荷块分布的影响,所以这里需要对丝氨酸S以及苏氨酸T设置2个dict#但是正电荷没有考虑到组氨酸H,所以这里没有设置组氨酸的正电荷phospho_charges = { 'S': -2, 'T':-2}#这个PTM翻译后修饰的字典可以根据实际情况进行修改,进行扩展sequence = sequence.upper()# 将序列转换为大写,以确保一致性charge_distribution = []# 初始化一个空列表来存储每个窗口的净电荷for i in range(len(sequence) - window_size + 1):# 遍历的窗口编号,从0开始window = sequence[i:i + window_size]# 获取当前窗口的序列total_charge = 0# 初始化当前窗口的总电荷for j, aa in enumerate(window):# enumerate(iterable, start=0),用于在遍历可迭代对象(如列表、字符串等)时,同时获取元素的索引和值,索引默认从0开始# j是索引从0开始,aa是氨基酸字符pos = i+j+1 #对应氨基酸的真实物理位置1-based,注意i是range从0开始(0:第1个窗口,j是enumrate返回的元组的索引,从0开始),所以pos=i+j+1#编号i=0的窗口、索引j=0的序列,实际上是第1个氨基酸pos=1charge = charges.get(aa, 0)# 需要计算电荷的氨基酸都在charges字典里,如果不在字典里,返回0if pos in phospho_sites:charge = phospho_charges.get(aa,0)#如果当前氨基酸在磷酸化位点列表里,则用磷酸化后的电荷替换掉原来的电荷total_charge += charge# 将氨基酸的电荷加到当前窗口的总电荷中charge_distribution.append(total_charge)# 将当前窗口的总电荷添加到charge_distribution列表中return charge_distribution# 返回电荷分布列表# 示例用法:
sequence = "KTTKIACKSPQPDPVDTPASTKQRPKRNLRKADVEEEFLALRKRTPSAGKAMDTPKPAVSDEKNINTFVETPVQKLDLLGNLPGSKRQPQTPKEKAEALEDLVGFKELFQTP" #替换为你的氨基酸序列
phospho_sites = [] #替换为你的磷酸化位点
charge_distribution = calculate_charge_distribution(sequence, phospho_sites)
print(charge_distribution)#绘制图形,需要用到matplotlib库
import matplotlib.pyplot as plt
plt.plot(charge_distribution)
plt.xlabel("Residue")
plt.ylabel("Charge")
plt.title("Charge plot")
plt.show()如果窗口是35内计算的净电荷,那R12:


如果是25窗口大小

很显然不对,因为RD12序列就长112,如果是使用窗口的话,实际上是无法延伸到100多位置的氨基酸序列的
所以实际上都和实物图对不上
其实看下面这张图就很清晰了:
这里是105到250都有,所以肯定不是这么画的
——》如果只是单纯看实物图的话,其实实物图肯定是每一个氨基酸位置都考虑进去的,
并且每个位置上的氨基酸序列肯定是计算了不止一个电荷的(所以x轴肯定又不是表征的单个氨基酸序列位置的信息,应该是某个集合或者是窗口下的位置信息之和)
问题就在于如果考虑了所有窗口内的氨基酸序列之后,
那几张又如何计算整个窗口下的
——》索性查看引用这篇NCB的所有文献里的charge plot怎么画的:




https://www.bioinformatics.nl/cgi-bin/emboss/charge
如果我只是输入同样的R12序列,结果是:

确实是能够计算到每一个位置,且电荷显然不是只计算一个,既然肯定有方法计算



我们重点关注的是下面NCPR的图,

we calculated the net charge per residue (NCPR) using a sliding window of 10 residues



没有关于电荷的图,但是倒是有一个有趣的图



总之我们能够看到,计算电荷块,看的多是整体区域的带电性质,也就是NPCR,一般不会用来看DE或者KRH数目的;
所以应该先搞清楚NCPR概念,然后再绘制charge plot(有一个参考来源即可)
参考2010的PNAS:



f+定义为一段给定aa序列中正电荷残基比例(理论上比例应该是这样算的:
f+=(带正电荷残基数x该残基所带正电荷电荷数)/该aa序列长度,
同理f-,
因为KRH和DE都是单位带电荷数,所以实际上就是看aa数目比例
以下图第一段序列为例:
全长29,正电荷氨基酸数9,负电荷为3,所以
f+=9/29=0.31
f-=3/29=0.10
其实就是将正负电荷均分到窗口/全长序列的每个残基中,比如说第一段序列相当于每个残基有31%的正电,同时带10%的负电,净电荷就是21%的正电荷

对应很多文献滑动窗口下的charge曲线实际上有正有负,定义NCPR=f±f-
(2)如何由电荷分布charge plot推导出电荷块charge block:
实际上就是如何表征电荷块
A charge block was designated when the area of the charge plot (window size: 35 amino acids) was larger than 20.
然后单个净电荷如何应用于计算电荷块:
设置一个窗口大小为35aa,但是是什么大于20呢:
带净电荷的aa数目?不合理,万一正的10个,负的10个 55开呢?
sum之后的净电荷值(应该是绝对值)?合理——》进一步评估
(3)必要时可可以进行多序列比对:
用的还是Clustal Omega( https://www.ebi.ac.uk/Tools/msa/clustalo/ )
(4)自定义的电荷模式参数:charge patterning parameters
文献里最终使用的是Blc参数

Dseg:

Blc:


另外还可以看一下reporting summary,实际上看到使用的数据分析方面的工具都是一些普通的python库

一些更细节的地方可以深入分析:一些问题都被peer提出来了

2,电荷模式参数计算:



实际使用参数评估过程中有很多参数可以使用:
kappa参数
SCD参数
Dseg参数
Blc参数

==================================================================
鉴于NCB文献中对于block的模糊定义,还是借鉴cell文献的method:


NCPR依据上面的定义我们能算了,然后窗口就是开10为大小,


window=10,如果NCPR>=5,则为酸性/负电block;
NCPR<=5,则为碱性/正电block
魔改一下我之前写的calc_NCR.py程序
import sys #提供参数
import csv #处理分隔符文件,不一定是csv
import argparse #用于参数解析,帮助手册
import subprocess #用于执行shell命令
# import os #用于文件操作# 定义一个函数来解析FASTA文件,主要是获得1个蛋白质的ID和氨基酸序列对应的字典sequences
def parse_fasta(fasta_file): with open(fasta_file, 'r') as file:#使用with语句打开文件,'r'表示以只读模式打开sequences = {} current_id = Nonecurrent_sequence = []#初始化了一个空字典sequences来存储蛋白质序列:一个变量current_id来存储当前处理的蛋白质的ID,以及一个列表current_sequence来存储当前蛋白质的氨基酸序列for line in file:line = line.strip()#遍历文件中的每一行,并使用strip()方法去除每行的换行符,这样后面append的时候所有序列会连在一起if line.startswith('>'):#检查当前行是否以'>'字符开头,如果是,那么这一行就是一个新的蛋白质记录的开始if current_id:sequences[current_id] = ''.join(current_sequence)current_sequence = []#如果current_id不为空(即之前已经处理过一个蛋白质),那么将当前蛋白质的序列(current_sequence)添加到sequences字典中,并重置current_sequence列表parts = line[1:].split()#对于每一行,从">"之后开始切割,然后获取['sp|Q6ZN18|AEBP2_HUMAN', 'Zinc', 'finger', 'protein', 'AEBP2', 'OS=Homo', 'sapiens', 'OX=9606', 'GN=AEBP2', 'PE=1', 'SV=2']id_info = parts[0].split('|')current_id = id_info[2] #实际上获取的sp|Q6ZN18|AEBP2_HUMAN中第3个分隔符字段才是对应的蛋白质名字,当然也可以对照GN字段后面的基因名字else:current_sequence.append(line)#对应前面的if,如果当前行不是以'>'字符开头,那么这一行就是一个蛋白质的氨基酸序列,将其添加到current_sequence列表中,直接添加并合并if current_id:sequences[current_id] = ''.join(current_sequence)#确保当前序列已经有一个ID(即 current_id 不为空,然后直接存储对应的序列到字典中return sequencesdef sliding_window_for_charged_block(sequence, phospho_sites=[],window_size=10, threshold=5):"""滑动窗口算法,用于查找蛋白质序列中NCPR绝对值大于阈值的窗口Args:sequence (str): 蛋白质序列phospho_sites (list): 磷酸化位点列表,默认为空,因为main主函数中是批量化处理,但显然每一个蛋白质序列都有差异的磷酸化位点,如果要分开计算,这个函数模块可以保留下来,但需要修改main函数window_size (int): 窗口大小,默认为10threshold (float): NCPR阈值,默认为5Returns:list: 合并后的窗口起点和终点列表,即找出的block"""charges = {'R': 1, 'K': 1,'H':1, 'E': -1, 'D': -1, 'S': 0, 'T': 0} #因为原文考虑到了丝氨酸以及苏氨酸的磷酸化与否对蛋白质电荷块分布的影响,所以这里需要对丝氨酸S以及苏氨酸T设置2个dict#NCB文献里正电荷没有考虑到组氨酸H,但是cell里有,还是补上phospho_charges = { 'S': -2, 'T':-2}#这个PTM翻译后修饰的字典可以根据实际情况进行修改,进行扩展results_acidic = [] #用于保存找到的酸性block的起点和终点,结果起点终点对保存在列表中results_basic = [] #用于保存找到的碱性block的起点和终点for i in range(len(sequence) - window_size + 1):# 遍历的窗口编号,从0开始 window = sequence[i:i + window_size]# 获取当前窗口的序列,注意取的是str切片,终点取不到,但是终点-起点+1-1total_charge = 0# 初始化当前窗口的总电荷for j, aa in enumerate(window):# enumerate(iterable, start=0),用于在遍历可迭代对象(如列表、字符串等)时,同时获取元素的索引和值,索引默认从0开始# j是索引从0开始,aa是氨基酸字符pos = i+j+1 #对应氨基酸的真实物理位置1-based,注意i是range从0开始(0:第1个窗口,j是enumrate返回的元组的索引,从0开始),所以pos=i+j+1#编号i=0的窗口、索引j=0的序列,实际上是第1个氨基酸pos=1charge = charges.get(aa, 0)# 需要计算电荷的氨基酸都在charges字典里,如果不在字典里,返回0if pos in phospho_sites:charge = phospho_charges.get(aa,0)#如果当前氨基酸在磷酸化位点列表里,则用磷酸化后的电荷替换掉原来的电荷total_charge += charge# 计算窗口内的总电荷if abs(total_charge) > threshold:# 窗口内的绝对值大于阈值,则判定为charged blockif total_charge > 0:results_basic.append((i+1, i + window_size))# 该起点终点对保存在正电碱性block列表中,(i+1, i + window_size)保存的数据类型是元组tuple,且是实际1-based的physical位置else:results_acidic.append((i+1, i + window_size))# 该起点终点对保存在酸性block列表中,(i+1, i + window_size)保存的数据类型是元组tuple# 合并重叠的窗口merged_results_acidic = []merged_results_basic =[] #同前面,对酸性负电以及碱性正电氨基酸的窗口进行合并,设置初始化的空列表# 用于保存已经合并的窗口起点终点元组对,注意每次比较都是比较当前元组的起点和上一个已经合并的元组(也就是merged_results[-1])的终点#下面是合并酸性带负电的窗口if results_acidic:# 如果酸性负电窗口results列表不为空merged_results_acidic = [results_acidic[0]]# 将第一个窗口加入merged_results列表中,作为后续比对的起点for current in results_acidic[1:]:# 遍历results_acidic列表中剩余的窗口last = merged_results_acidic[-1]# 取出已经合并的窗口的最后一个窗口的元组对if current[0] <= last[1] + 1: # 如果当前窗口与最后一个窗口有重叠,也就是当前窗口的起点小于等于已经合并的最后一个窗口的终点+1(overlap以及相邻)merged_results_acidic[-1] = (last[0], max(last[1], current[1]))# 更新已经合并的最后一个窗口的终点为当前窗口和已经合并的最后一个窗口的终点的最大值,总之就是更新最后一个窗口,起点不变,终点更新else:merged_results_acidic.append(current)# 如果当前窗口与最后一个窗口没有重叠,直接将当前窗口加入merged_results列表中,也就是当前窗口并没有重叠#下面是合并碱性带正电的窗口if results_basic:merged_results_basic = [results_basic[0]]for current in results_basic[1:]:last = merged_results_basic[-1]if current[0] <= last[1] + 1:merged_results_basic[-1] = (last[0], max(last[1], current[1]))else:merged_results_basic.append(current)return merged_results_acidic, merged_results_basic# 返回合并之后的酸性负电窗口和碱性正电窗口,各自是合并后的窗口起点和终点列表,且每一个元素是元组,后续需要解包赋值def main(fasta_file, output_file,window_size, threshold, delimiter='\t'): #默认输出为tsv格式sequences = parse_fasta(fasta_file)#调用parse_fasta函数来解析FASTA文件,并返回一个字典sequences,其中键是蛋白质的ID,值是蛋白质的氨基酸序列(字符串)#处理正电荷block、负电荷block区域输出文件with open(output_file, 'w') as csvfile:#使用with语句打开一个输出文件(需要自己提供参数命名,'w'表示以写入模式打开writer = csv.writer(csvfile, delimiter=delimiter)#创建一个csv.writer对象,用于将数据写入csv文件,delimiter='\t'表示使用制表符作为字段分隔符writer.writerow(['Protein_Name', 'charged_block_start', 'charged_block_end', 'Sequence', 'charged_block_type'])#写入表头,即列名for protein_name, sequence in sequences.items():#sequences是一个字典,键是蛋白质的ID,值是蛋白质的氨基酸序列,现在从值中获取charged block区域起点以及终点的元组对列表merged_results_acidic, merged_results_basic= sliding_window_for_charged_block(sequence.replace('\n', ''),phospho_sites=[],window_size=window_size, threshold=threshold) #.replace('\n', '')双重保证,如果前面fasta序列中没有去除换行符for results in merged_results_acidic:writer.writerow([protein_name, results[0], results[1], sequence[results[0]-1:results[1]], 'Acidic'])for results in merged_results_basic:writer.writerow([protein_name, results[0], results[1], sequence[results[0]-1:results[1]], 'Basic'])#将蛋白质的ID、charged block区域的起点和终点,以及对应的序列切片,以及是酸性还是碱性,写入csv文件中,注意切片是0-based的,而真实位置是1-based的#后续可以导入到R中使用tidyverse#如果需要统计信息并写入到{output_file}.stats文件中,可以参考我原本calc_NCR.py的代码,这里不再赘述 #使用 argparse 模块来实现类似于 Linux 工具的命令行参数解析(包括生成帮助手册等)if __name__ == "__main__":parser = argparse.ArgumentParser(description="FASTA文件处理工具")parser.add_argument("--fasta_file", help="输入的FASTA文件路径")parser.add_argument("--delimiter", help="输出文件的分隔符(如$'\t'等)")parser.add_argument("--window_size", type=int, default=10, help="滑动窗口大小")parser.add_argument("--threshold", type=float, default=5, help="NCPR阈值,注意是>=")parser.add_argument("--output_file", help="输出可变电荷块文件路径")# parser.add_argument("--help", action="help", help="显示帮助信息"),不需要手动添加 --help 选项,因为 argparse 模块已经自动添加了一个 --help 选项来显示帮助信息qargs = parser.parse_args()main(args.fasta_file, args.output_file, args.delimiter, args.window_size, args.threshold)
去掉符号修改之后是
import sys #提供参数
import csv #处理分隔符文件,不一定是csv
import argparse #用于参数解析,帮助手册
import subprocess #用于执行shell命令
# import os #用于文件操作# 定义一个函数来解析FASTA文件,主要是获得1个蛋白质的ID和氨基酸序列对应的字典sequences
def parse_fasta(fasta_file): with open(fasta_file, 'r') as file:#使用with语句打开文件,'r'表示以只读模式打开sequences = {} current_id = Nonecurrent_sequence = []#初始化了一个空字典sequences来存储蛋白质序列:一个变量current_id来存储当前处理的蛋白质的ID,以及一个列表current_sequence来存储当前蛋白质的氨基酸序列for line in file:line = line.strip()#遍历文件中的每一行,并使用strip()方法去除每行的换行符,这样后面append的时候所有序列会连在一起if line.startswith('>'):#检查当前行是否以'>'字符开头,如果是,那么这一行就是一个新的蛋白质记录的开始if current_id:sequences[current_id] = ''.join(current_sequence)current_sequence = []#如果current_id不为空(即之前已经处理过一个蛋白质),那么将当前蛋白质的序列(current_sequence)添加到sequences字典中,并重置current_sequence列表parts = line[1:].split()#对于每一行,从">"之后开始切割,然后获取['sp|Q6ZN18|AEBP2_HUMAN', 'Zinc', 'finger', 'protein', 'AEBP2', 'OS=Homo', 'sapiens', 'OX=9606', 'GN=AEBP2', 'PE=1', 'SV=2']id_info = parts[0].split('|')current_id = id_info[2] #实际上获取的sp|Q6ZN18|AEBP2_HUMAN中第3个分隔符字段才是对应的蛋白质名字,当然也可以对照GN字段后面的基因名字else:current_sequence.append(line)#对应前面的if,如果当前行不是以'>'字符开头,那么这一行就是一个蛋白质的氨基酸序列,将其添加到current_sequence列表中,直接添加并合并if current_id:sequences[current_id] = ''.join(current_sequence)#确保当前序列已经有一个ID(即 current_id 不为空,然后直接存储对应的序列到字典中return sequencesdef sliding_window_for_charged_block(sequence, phospho_sites=[],window_size=10, threshold=5):"""滑动窗口算法,用于查找蛋白质序列中NCPR绝对值大于阈值的窗口Args:sequence (str): 蛋白质序列phospho_sites (list): 磷酸化位点列表,默认为空,因为main主函数中是批量化处理,但显然每一个蛋白质序列都有差异的磷酸化位点,如果要分开计算,这个函数模块可以保留下来,但需要修改main函数window_size (int): 窗口大小,默认为10threshold (float): NCPR阈值,默认为5Returns:list: 合并后的窗口起点和终点列表,即找出的block"""charges = {'R': 1, 'K': 1,'H':1, 'E': -1, 'D': -1, 'S': 0, 'T': 0} #因为原文考虑到了丝氨酸以及苏氨酸的磷酸化与否对蛋白质电荷块分布的影响,所以这里需要对丝氨酸S以及苏氨酸T设置2个dict#NCB文献里正电荷没有考虑到组氨酸H,但是cell里有,还是补上phospho_charges = { 'S': -2, 'T':-2}#这个PTM翻译后修饰的字典可以根据实际情况进行修改,进行扩展results_acidic = [] #用于保存找到的酸性block的起点和终点,结果起点终点对保存在列表中results_basic = [] #用于保存找到的碱性block的起点和终点for i in range(len(sequence) - window_size + 1):# 遍历的窗口编号,从0开始 window = sequence[i:i + window_size]# 获取当前窗口的序列,注意取的是str切片,终点取不到,但是终点-起点+1-1total_charge = 0# 初始化当前窗口的总电荷for j, aa in enumerate(window):# enumerate(iterable, start=0),用于在遍历可迭代对象(如列表、字符串等)时,同时获取元素的索引和值,索引默认从0开始# j是索引从0开始,aa是氨基酸字符pos = i+j+1 #对应氨基酸的真实物理位置1-based,注意i是range从0开始(0:第1个窗口,j是enumrate返回的元组的索引,从0开始),所以pos=i+j+1#编号i=0的窗口、索引j=0的序列,实际上是第1个氨基酸pos=1charge = charges.get(aa, 0)# 需要计算电荷的氨基酸都在charges字典里,如果不在字典里,返回0if pos in phospho_sites:charge = phospho_charges.get(aa,0)#如果当前氨基酸在磷酸化位点列表里,则用磷酸化后的电荷替换掉原来的电荷total_charge += charge# 计算窗口内的总电荷if abs(total_charge) >= threshold:# 窗口内的绝对值大于等于阈值,则判定为charged blockif total_charge > 0:results_basic.append((i+1, i + window_size))# 该起点终点对保存在正电碱性block列表中,(i+1, i + window_size)保存的数据类型是元组tuple,且是实际1-based的physical位置else:results_acidic.append((i+1, i + window_size))# 该起点终点对保存在酸性block列表中,(i+1, i + window_size)保存的数据类型是元组tuple# 合并重叠的窗口merged_results_acidic = []merged_results_basic =[] #同前面,对酸性负电以及碱性正电氨基酸的窗口进行合并,设置初始化的空列表# 用于保存已经合并的窗口起点终点元组对,注意每次比较都是比较当前元组的起点和上一个已经合并的元组(也就是merged_results[-1])的终点#下面是合并酸性带负电的窗口if results_acidic:# 如果酸性负电窗口results列表不为空merged_results_acidic = [results_acidic[0]]# 将第一个窗口加入merged_results列表中,作为后续比对的起点for current in results_acidic[1:]:# 遍历results_acidic列表中剩余的窗口last = merged_results_acidic[-1]# 取出已经合并的窗口的最后一个窗口的元组对if current[0] <= last[1] + 1: # 如果当前窗口与最后一个窗口有重叠,也就是当前窗口的起点小于等于已经合并的最后一个窗口的终点+1(overlap以及相邻)merged_results_acidic[-1] = (last[0], max(last[1], current[1]))# 更新已经合并的最后一个窗口的终点为当前窗口和已经合并的最后一个窗口的终点的最大值,总之就是更新最后一个窗口,起点不变,终点更新else:merged_results_acidic.append(current)# 如果当前窗口与最后一个窗口没有重叠,直接将当前窗口加入merged_results列表中,也就是当前窗口并没有重叠#下面是合并碱性带正电的窗口if results_basic:merged_results_basic = [results_basic[0]]for current in results_basic[1:]:last = merged_results_basic[-1]if current[0] <= last[1] + 1:merged_results_basic[-1] = (last[0], max(last[1], current[1]))else:merged_results_basic.append(current)return merged_results_acidic, merged_results_basic# 返回合并之后的酸性负电窗口和碱性正电窗口,各自是合并后的窗口起点和终点列表,且每一个元素是元组,后续需要解包赋值def main(fasta_file, output_file,window_size, threshold): #默认输出为tsv格式sequences = parse_fasta(fasta_file)#调用parse_fasta函数来解析FASTA文件,并返回一个字典sequences,其中键是蛋白质的ID,值是蛋白质的氨基酸序列(字符串)#处理正电荷block、负电荷block区域输出文件with open(output_file, 'w') as csvfile:#使用with语句打开一个输出文件(需要自己提供参数命名,'w'表示以写入模式打开writer = csv.writer(csvfile, delimiter='\t')#创建一个csv.writer对象,用于将数据写入csv文件,delimiter='\t'表示使用制表符作为字段分隔符writer.writerow(['Protein_Name', 'charged_block_start', 'charged_block_end', 'Sequence', 'charged_block_type'])#写入表头,即列名for protein_name, sequence in sequences.items():#sequences是一个字典,键是蛋白质的ID,值是蛋白质的氨基酸序列,现在从值中获取charged block区域起点以及终点的元组对列表merged_results_acidic, merged_results_basic= sliding_window_for_charged_block(sequence.replace('\n', ''),phospho_sites=[],window_size=window_size, threshold=threshold) #.replace('\n', '')双重保证,如果前面fasta序列中没有去除换行符for results in merged_results_acidic:writer.writerow([protein_name, results[0], results[1], sequence[results[0]-1:results[1]], 'Acidic'])for results in merged_results_basic:writer.writerow([protein_name, results[0], results[1], sequence[results[0]-1:results[1]], 'Basic'])#将蛋白质的ID、charged block区域的起点和终点,以及对应的序列切片,以及是酸性还是碱性,写入csv文件中,注意切片是0-based的,而真实位置是1-based的#后续可以导入到R中使用tidyverse#如果需要统计信息并写入到{output_file}.stats文件中,可以参考我原本calc_NCR.py的代码,这里不再赘述 #使用 argparse 模块来实现类似于 Linux 工具的命令行参数解析(包括生成帮助手册等)if __name__ == "__main__":parser = argparse.ArgumentParser(description="接受uniprot输入的蛋白质原始fasta序列文件(可以是一群蛋白质),计算每个窗口下的NCPR值,输出NCPR值大于等于阈值的窗口(也就是酸性带正电氨基酸block,或碱性带负电氨基酸block)的起点和终点")parser.add_argument("--fasta_file", help="输入的蛋白质FASTA文件路径")# parser.add_argument("--delimiter", help="输出文件的分隔符(如$'\\t'等),默认是制表符", default='\t')parser.add_argument("--window_size", type=int, default=10, help="滑动窗口大小,默认是10")parser.add_argument("--threshold", type=float, default=5, help="NCPR阈值,默认是5,注意是>=")parser.add_argument("--output_file", help="输出可变电荷块文件路径")# parser.add_argument("--help", action="help", help="显示帮助信息"),不需要手动添加 --help 选项,因为 argparse 模块已经自动添加了一个 --help 选项来显示帮助信息qargs = parser.parse_args()main(args.fasta_file, args.output_file, args.window_size, args.threshold)

python3 calc_charged_block.py --fasta_file /mnt/sdb/zht/project/uniprot/uniprot_870.fasta --window_size 10 --threshold 5 --output_file uniprot_870_NCPR_10_5.tsv
大概效果如下:

实际上是可以和IDR等各种bed区域信息都统一起来
下一步就是结构预测,或者是提取感兴趣的区域做个结构表位分析(motif,奇美拉/pymol中搜索motif表征之类的)
相关文章:
NCR+可变电荷块3——NCB/cell绘图1
文献method参考: 蛋白质序列数据从uniprot中获取 https://www.uniprot.org/uniprotkb/P46013/entry https://www.uniprot.org/uniprotkb/P06748/entry、 1,电荷分布计算: Charge distribution was calculated as the sum of the charges …...

数据仓库是什么?数据仓库简介
数据仓库(Data Warehouse)是一个面向主题的、集成的、相对稳定的、反映历史变化的数据集合,用于支持企业的管理决策。以下是对数据仓库的详细解释: 一、定义与特性 定义:数据仓库是构建在组织的现有数据基础上&#x…...

AI的进阶之路:从机器学习到深度学习的演变(二)
AI的进阶之路:从机器学习到深度学习的演变(一) 三、机器学习(ML):AI的核心驱动力 3.1 机器学习的核心原理 机器学习(Machine Learning, ML)突破了传统编程的局限,它不再…...
)
C++中属性(Attributes)
属性(Attributes)在 C 中的完整讲解 在 C 中,属性(Attributes) 是一种编译时机制,用于附加元数据到函数、变量、类型等元素上,以指导编译器如何优化、检查、警告或者改变编译行为。通过属性&am…...
Go语言中的defer,panic,recover 与错误处理
目录 前言 三个关键字 defer语句 panic语句 recover函数 defer、panic、recover组成的错误处理 总结 前言 在其他编程语言中,如Java,宕机往往以异常的形式存在。底层抛出异常,上层逻辑通过try...catch...fanally机制捕获异常并处理&am…...
力扣 904.水果成篮)
(C语言)力扣 904.水果成篮
写在所有的前面: 本文采用C语言实现代码 目录 写在所有的前面:题目说明题目:力扣 904.水果成篮题目出处题目描述Description输入Input输出Output样例Sample限制Hint 解答说明方案解题思路一般情况特殊情况 代码实现其他解释 题目说明 题目…...

2024 年12月英语六级CET6听力原文(Lecture部分)
2024 年12月英语六级CET6听力原文(Long Conersation和Passage) 1 牛津大学关于普遍道德准则的研究及相关观点与建议 译文 2 食物颜色对味觉体验及大脑预期的影响 译文 3 财务资源对意义与幸福之间关系的影响研究 译文...

CentOS下,离线安装vscode的步骤;
前置条件: 1.CentOS7; 步骤: 1.下载vscode指定版本,例如; 例如 code-1.83.1-1696982959.el7.x86_64.rpm 2.使用下面命令: sudo rpm -ivh code-1.83.1-1696982959.el7.x86_64.rpm 其他: 卸载vscode的命…...

ubuntu停止.netcore正在运行程序的方法
在Ubuntu系统中停止正在运行的.NET Core程序,你可以使用以下几种方法: 使用kill命令: 如果你知道.NET Core程序的进程ID(PID),你可以直接使用kill命令来停止它。首先,使用ps命令配合grep来查找.…...

机器学习基础 衡量模型性能指标
目录 1 前言 编辑1.1 错误率(Error rate)&精度(Accuracy)&误差(Error): 1.2 过拟合(overfitting): 训练误差小,测试误差大 1.3 欠拟合(underfitting):训练误差大,测试误差大 1.4 MSE: 1.5 RMSE: 1.6 MAE: 1.7 R-S…...
及形态学处理)
《OpenCV计算机视觉》-对图片的各种操作(均值、方框、高斯、中值滤波处理)及形态学处理
文章目录 《OpenCV计算机视觉》-对图片的各种操作(均值、方框、高斯、中值滤波处理)边界填充阈值处理图像平滑处理生成椒盐图片均值滤波处理方框滤波处理高斯滤波处理中值滤波处理 图像形态学腐蚀膨胀开运算闭运算顶帽和黑帽 《OpenCV计算机视觉》-对图片…...

如何让Tplink路由器自身的IP网段 与交换机和电脑的IP网段 保持一致?
问题分析: 正常情况下,我的需求是:电脑又能上网,又需要与路由器处于同一局域网下(串流Pico4 VR眼镜),所以,我是这么连接 交换机、路由器、电脑 的: 此时,登录…...

【JetPack】Navigation知识点总结
Navigation的主要元素: 1、Navigation Graph: 一种新的XML资源文件,包含应用程序所有的页面,以及页面间的关系。 <?xml version"1.0" encoding"utf-8"?> <navigation xmlns:android"http://schemas.a…...

InnoDB引擎的内存结构
InnoDB擅长处理事务,具有自动崩溃恢复的特性 架构图: 由4部分组成: 1.Buffer Pool:缓冲池,缓存表数据和索引数据,减少磁盘I/O操作,提升效率 2.change Buffer:写缓冲区,…...

Y3地图制作1:水果缤纷乐、密室逃脱
文章目录 一、水果缤纷乐1.1 游戏设计1.1.1 项目解析1.1.2 项目优化1.1.3 功能拆分 1.2 场景制作1.2.1 场景需求1.2.2 创建主镜头、绘制草稿,构思文案和情景1.2.3 构建场景地图1.2.4 光源与氛围设置 1.3 游戏初始化1.3.1 物编、UI预设置1.3.2 游戏初始化1.3.2 玩家初…...

ESP32_H2(IDF)学习系列-ADC模数转换(连续转换)
一、简介(节选手册) 1 概述 ESP32-H2 搭载了以下模拟外设: • 一个 12 位逐次逼近型模拟数字转换器 (SAR ADC),用于测量最多来自 5 个管脚上的模拟信号。 • 一个温度传感器,用于测量及监测芯片内部温度。 2 SAR ADC 2…...

如何通过TikTok成功引流到独立站
随着短视频平台的迅猛发展,TikTok已成为全球最受欢迎的社交媒体之一,尤其是在年轻用户群体中更是势不可挡。如果你是一个独立站(如电商网站、博客、个人品牌站等)的运营者,那么如何通过TikTok引流到独立站已经成为一个…...

生成签名文件 .keystore
打开java sdk 到bin目录(D:\JDK\Java\jdk1.8.0_202\bin),打开dos窗口执行以下命令: 命令行输入: 1、生成签名文件:(-alias 别名 validity 有效期 9125 天) keytool -genkeypair -v…...

Mono里运行C#脚本3—mono_jit_init
前面已经介绍了配置参数的读取,这样就可以把一些特殊的配置读取进来,完成了用户配置阶段的参数,接着下来就需要进行大工程的建造了。 为什么这样说呢,因为需要解释并执行C#编译的受托管的代码,相当于就是建立一个C#代码运行的虚拟机,而这个虚拟机还是很复杂的,不但要支…...
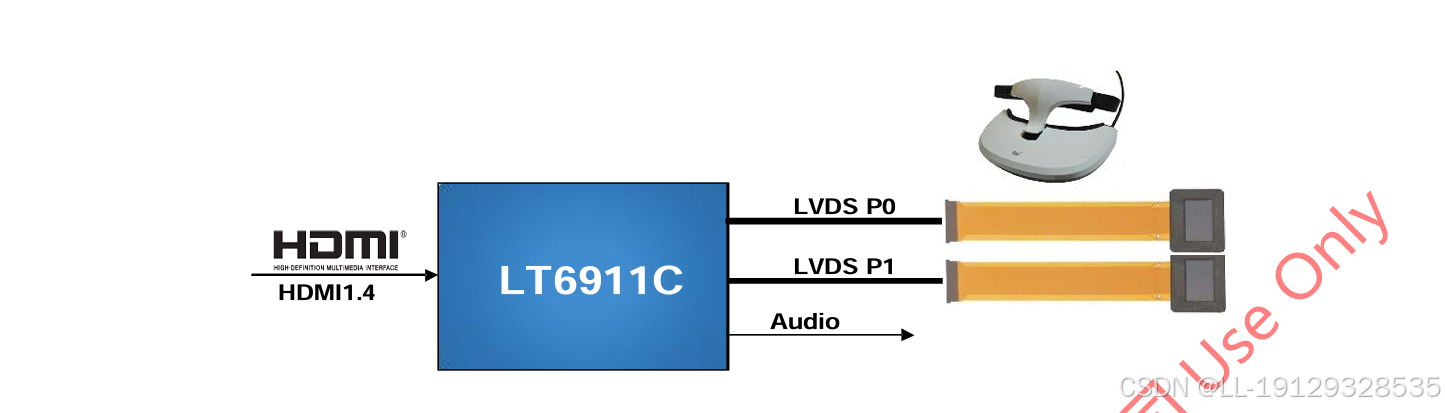
龙迅#LT6911C适用于HDMI转MIPI/LVDS产品应用,分辨率高达4K30HZ,内置程序,支持KEY(HDCP)!
1. 描述 LT6911C 是一款高性能 HDMI1.4/DP 转 MIPIDSI/CSI/LVDS 芯片,适用于 VR/智能手机/显示应用。 对于 MIPIDSI/CSI 输出,LT6911C具有可配置的单端口或双端口 MIPIDSI/CSI,具有 1 个高速时钟通道和 1~4 个高速数据通道,运行…...

Qwen3.5-2B本地知识库问答系统:基于CSDN技术文章的精准检索与摘要
Qwen3.5-2B本地知识库问答系统:基于CSDN技术文章的精准检索与摘要 1. 技术问答的痛点与解决方案 技术开发者在日常工作中经常遇到这样的场景:遇到一个具体的技术问题,需要快速找到相关解决方案。传统的做法是在搜索引擎中输入关键词&#x…...

UR机械臂ROS2驱动选型指南:深入对比Ethernet RTDE与EtherCAT,你的项目该怎么选?
UR机械臂ROS2驱动选型指南:Ethernet RTDE与EtherCAT深度对比与实战决策 在工业自动化与协作机器人领域,UR(Universal Robots)机械臂因其灵活性和易用性广受青睐。然而,当工程师们将UR机械臂集成到ROS2生态系统中时&…...

AI驱动的下一代云ERP:SAP Cloud ERP 2602 更新亮点小结
大家好,SAP Cloud ERP 2602版本更新了!2602的一个核心特点,是在保持标准化 SaaS 的前提下,将“嵌入式 AI 自然语言交互 Agentic AI”有机结合,让用户可以在熟悉的业务流程中,以对话方式完成信息查询、数据…...

webpack优化:Vue配置compression-webpack-plugin实现gzip压缩
需求实现 1.安装依赖 npm i -D compression-webpack-plugin6.1.12.修改vue .config.js配置 const CompressionPlugin require(compression-webpack-plugin) // gzip 相关 const isGZIP process.env.VUE_APP_GZIP ONmodule.exports {configureWebpack(config) {if (isGZ…...

3步构建安全可靠的用户脚本生态系统:Greasy Fork深度技术解析
3步构建安全可靠的用户脚本生态系统:Greasy Fork深度技术解析 【免费下载链接】greasyfork An online repository of user scripts. 项目地址: https://gitcode.com/gh_mirrors/gr/greasyfork Greasy Fork作为开源的用户脚本平台,基于Ruby on Rai…...

DockerUI仪表板定制终极指南:7步打造个性化监控界面
DockerUI仪表板定制终极指南:7步打造个性化监控界面 【免费下载链接】ui-for-docker A web interface for Docker, formerly known as DockerUI. This repo is not maintained 项目地址: https://gitcode.com/gh_mirrors/ui/ui-for-docker DockerUI是一个基于…...

颠覆传统:5大核心技术让百度网盘提取码获取效率提升10倍
颠覆传统:5大核心技术让百度网盘提取码获取效率提升10倍 【免费下载链接】baidupankey 项目地址: https://gitcode.com/gh_mirrors/ba/baidupankey 在数字化资源交互日益频繁的今天,百度网盘作为国内主流的文件分享平台,其提取码机制…...

SpringBoot的生命周期原理分析之一SpringBoot准备容器与环境
目录 1.SpringBootApplication准备 1.1SpringApplication创建 1.2.1保存主配置源 1.2.2推断Web环境 1.2.3设置初始化器 1.2.4设置监听器 1.2.5确定主启动类 1.2.6扩展了解:SpringBoot的发展 1.2SpringBootApplication启动 1.2.1启动计时与全局异常处理机制…...

Step3-VL-10B与Keil5开发环境:嵌入式视觉系统实战
Step3-VL-10B与Keil5开发环境:嵌入式视觉系统实战 用最简单的方式,带你从零搭建一个能"看懂世界"的嵌入式视觉系统 1. 开篇:为什么需要嵌入式视觉? 你有没有想过,让一个小小的单片机也能像人一样"看见…...

零基础玩转Qwen-Image-2512-SDNQ-uint4-svd-r32:Web界面一键生成图片
零基础玩转Qwen-Image-2512-SDNQ-uint4-svd-r32:Web界面一键生成图片 1. 快速了解Qwen-Image-2512-SDNQ-uint4-svd-r32 Qwen-Image-2512-SDNQ-uint4-svd-r32是一款基于Stable Diffusion技术的高性能图片生成模型,经过特殊优化后体积更小、运行更快。这…...