快速、准确地检测和分类病毒序列分析工具 ViralCC的介绍和详细使用方法, 附带应用脚本
介绍

viralcc是一个基因组病毒分析工具,可以用于快速、准确地检测和分类病毒序列。
github:dyxstat/ViralCC: ViralCC: leveraging metagenomic proximity-ligation to retrieve complete viral genomes (github.com)
Instruction of reproducing results in ViralCC paper:dyxstat/Reproduce_ViralCC: Instruction of reproducing results in ViralCC paper (github.com)
安装viralcc:
首先,确保你已经安装了Python 3.6或更高版本。
从GitHub上下载viralcc的代码。在终端中输入以下命令:
git clone https://github.com/dyxstat/ViralCC.git
进入viralcc文件夹:
cd viralcc
建议使用mamba 或 conda 直接安装吧:
#安装前先修改配置文件viralcc_linux_env.yaml,将环境名称修改为自己想要的
#其他的东西不要动name: viralcc //修改这个就行了,原来为ViralCC_ENV
channels:- bioconda- conda-forge- defaults- r
dependencies:- _libgcc_mutex=0.1- _openmp_mutex=4.5- _r-mutex=1.0.1- binutils_impl_linux-64=2.35.1- binutils_linux-64=2.35- biopython=1.78- bwidget=1.9.14- bzip2=1.0.8
mamba安装:
mamba env create -f viralcc_linux_env.yaml使用viralcc:
在终端中输入以下命令,可以查看viralcc的可用命令和选项:
mamba activate viralccpython ./viralcc.py -h
usage: viralcc.py [-h] {pipeline} ...ViralCC: a metagenomic proximity-based tool to retrieve complete viral genomesoptional arguments:-h, --help show this help message and exitcommands:Valid commands准备输入文件。viralcc支持FASTA和FASTQ格式的输入文件,你可以将你的病毒序列文件准备好。
运行viralcc进行病毒分析测试。在终端中输入以下命令:
python ./viralcc.py pipeline -v Test/final.contigs.fa Test/MAP_SORTED.bam Test/viral_contigs.txt Test/out_test使用分析流程:
指令:处理原始数据 按照本节的指示,对原始shotgun和Hi-C数据进行处理,并生成ViralCC的输入:
-
清理原始shotgun和Hi-C读段 使用BBTools套件中的bbduk工具去除接头序列,参数为ktrim=r k=23 mink=11 hdist=1 minlen=50 tpe tbo;同时使用bbduk进行质量修剪,参数为trimq=10 qtrim=r ftm=5 minlen=50。另外,通过设置bbduk参数ftl=10来剪切Hi-C读段的前10个核苷酸。使用BBTools套件中的clumpify.sh脚本来移除Hi-C读段中的相同PCR光学重复和Tile边缘重复。
-
组装shotgun读段 对shotgun文库,采用如MEGAHIT之类的de novo组装软件进行元基因组组装。
megahit -1 SG1.fastq.gz -2 SG2.fastq.gz -o ASSEMBLY --min-contig-len 1000 --k-min 21 --k-max 141 --k-step 12 --merge-level 20,0.95 -
将Hi-C双端读段比对到组装得到的contigs上 使用如BWA MEM这样的DNA比对软件将Hi-C双端读段比对至已组装的contigs。然后应用samtools(参数为‘view -F 0x904’)移除未比对、补充比对以及二级比对的读段。需要使用'samtools sort'按名称对BAM文件进行排序。
bwa index final.contigs.fa bwa mem -5SP final.contigs.fa hic_read1.fastq.gz hic_read2.fastq.gz > MAP.sam samtools view -F 0x904 -bS MAP.sam > MAP_UNSORTED.bam samtools sort -n MAP_UNSORTED.bam -o MAP_SORTED.bam -
从组装的contigs中识别病毒contigs 利用如VirSorter这样的病毒序列检测软件对组装后的contigs进行筛选以识别病毒contigs。
wrapper_phage_contigs_sorter_iPlant.pl -f final.contigs.fa --db 1 --wdir virsorter_output --data-dir virsorter-data
指令:运行ViralCC
python ./viralcc.py pipeline [参数] FASTA文件 BAM文件 VIRAL文件 输出目录参数说明: --min-len: 可接受的最小contig长度(默认值为1000) --min-mapq: 最小可接受的比对质量(默认值为30) --min-match: 接受的比对至少要有N个匹配(默认值为30) --min-k: 确定宿主邻近图的k值下限(默认值为4) --random-seed: Leiden聚类算法的随机种子(默认值为42) --cover (可选): 覆盖现有文件。如果不指定此选项,若检测到输出文件已存在,则会返回错误。 -v (可选): 显示有关ViralCC过程更多详细信息的详尽输出。
输入文件: FASTA_file: 已组装contig的fasta文件(例如:Test/final.contigs.fa) BAM_file: Hi-C比对结果的bam文件(例如:Test/MAP_SORTED.bam) VIRAL_file: 包含识别出的病毒contigs名称的txt文件,每行一个名称且无表头(例如:Test/viral_contigs.txt)
输出文件: VIRAL_BIN: 包含草稿病毒bin的fasta文件夹 cluster_viral_contig.txt: 聚类结果,包含两列,第一列是病毒contig名称,第二列是组号 viral_contig_info.csv: 病毒contig信息,包含三列(contig名称、contig长度和GC含量) prokaryotic_contig_info.csv: 非病毒contig信息,包含三列(contig名称、contig长度和GC含量) viralcc.log: ViralCC日志文件
示例:
python ./viralcc.py pipeline -v final.contigs.fa MAP_SORTED.bam viral_contigs.txt out_directory实用脚本位置:Reproduce_ViralCC/Scripts at main · dyxstat/Reproduce_ViralCC (github.com)
concatenation.py
import os
import io
import sys
import argparse
import Bio.SeqIO as SeqIO
import gzip
import numpy as np
import pandas as pddef get_no_hidden_folder_list(wd):folder_list = []for each_folder in os.listdir(wd):if not each_folder.startswith('.'):folder_list.append(each_folder)folder_list_sorte = sorted(folder_list)return folder_list_sortedef main(path , output_file):file_list = get_no_hidden_folder_list(path)bin_num = len(file_list) for k in range(bin_num):seq_file = '%s/%s' % (path , file_list[k])if k==0:op1 = 'echo ' + '\">BIN_' + str(k) + '\" ' + '> ' + output_fileelse:op1 = 'echo ' + '\">BIN_' + str(k) + '\" ' + '>> ' + output_fileos.system(op1)op2 = 'grep ' + '-v ' + '\'>\' ' + seq_file + ' >> ' + output_fileos.system(op2) if __name__ == "__main__":parser = argparse.ArgumentParser()parser.add_argument("-p",help="path")parser.add_argument("-o",help="output_file")args=parser.parse_args()main(args.p,args.o)find_viral_contig.R
virsorterfile = 'VIRSorter_global-phage-signal.csv'
vs.pred <- read.csv(virsorterfile,quote="",head=F)
vs.head <- read.table(virsorterfile,sep=",",quote="",head=T,comment="",skip=1,nrows=1)
colnames(vs.pred) <- colnames(vs.head)
colnames(vs.pred)[1] <- "vs.id"
vs.cats <- do.call(rbind,strsplit(x=as.character(vs.pred$vs.id[grep("category",vs.pred$vs.id)]),split=" - ",fixed=T))[,2]
vs.num <- grep("category",vs.pred$vs.id)
vs.pred$Category <- paste(c("",rep.int(vs.cats, c(vs.num[-1],nrow(vs.pred)) - vs.num)), vs.pred$Category)
vs.pred <- vs.pred[-grep("#",vs.pred$vs.id),]vs.pred$node <- gsub(pattern="VIRSorter_",replacement="",x=vs.pred$vs.id)
vs.pred$node <- gsub(pattern="-circular",replacement="",x=vs.pred$node)
vs.pred$node <- gsub(pattern="cov_(\\d+)_",replacement="cov_\\1.",x=vs.pred$node,perl=F)rownames(vs.pred) = seq(1 , 1393)vs_phage = vs.pred[1:1338 , ]phage_name = vs_phage$nodefor(i in 1:1338)
{temp = paste0(strsplit(phage_name[i],split='_')[[1]][1] , '_' , strsplit(phage_name[i],split='_')[[1]][2])phage_name[i] = temp
}group_name = rep('group0' , 1338)
phage = cbind(phage_name , group_name)write.table(phage , file = 'viral.txt' , sep='\t', row.names = F , col.names = F , quote =FALSE)plot_graph.R
####################write ggplot figure###############
library(ggplot2)
library(ggpubr)
library(ggforce)theme_set(theme_bw()+theme(panel.spacing=grid::unit(0,"lines")))##########柱状图对于不同方法和分类###########
Rank = rep(c('F-score' , 'ARI' , 'NMI' , 'Homogeneity') , each = 5)
Pipeline = rep(c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'),times = 4)
Number = c(0.198,0.485,0.366,0.404,0.795,0.111,0.471,0.302,0.274,0.787,0.724,0.742,0.782,0.817,0.929,0.570,0.723,0.687,0.691,0.921)col = c('#8FBC94' , '#4FB0C6', "#4F86C6", "#527F76", '#CC9966')df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))
df$Rank = factor(df$Rank , levels = c('F-score' , 'ARI' , 'NMI', 'Homogeneity'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'dodge')+scale_fill_manual(values = col,limits= c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))+coord_cartesian(ylim = c(0.05,0.975))+labs(x = "Clustering metrics", y = "Scores", title = "The mock human gut dataset")+theme(legend.position="bottom",legend.title=element_blank(),legend.text = element_text(size = 12),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("fig2a.eps", width = 7 , height = 6 , device = cairo_ps)Rank = rep(c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 5)
Number = c(2,4,1,1,5,5, 6,1,0,1,0,5, 4,2,26)col = c("#8FBC94","#77AAAD","#6E7783")df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'stack')+scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+labs(x = "Binning method", y = "Number of viral bins", title = "The mock human gut dataset")+theme(legend.position="bottom",legend.title=element_blank(),legend.text = element_text(size = 12),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("fig2b.eps", width = 7, height = 6, device = cairo_ps)viral_num = data.frame('number' = c(1, 4 , 1 , 1 , 13),'method' = c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))viral_num$method = factor(viral_num$method , levels=c('VAMB' , 'CoCoNet' , 'vRhyme' , 'bin3C' , 'ViralCC'))ggplot(data = viral_num, aes(x = method , y = number )) + geom_bar(stat = "identity", position='dodge' , width = 0.9,fill = 'steelblue') + labs(x = 'Binning method', y = 'Number of high-quality vMAGs within the co-host systems', title = "The mock human gut dataset") +theme(panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("fig2c.eps", width = 7, height = 6, device = cairo_ps)##############human gut 2a############
Rank = rep(c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'),each = 5)
Completeness = rep(c( "≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"),times = 5)
###Number needs to be 4*5 matrix##
Number = c(11 , 12 , 17 , 7 , 78,1 , 0 , 1 , 4 , 33,10, 11, 10, 6, 60,2, 1 , 3 , 2 , 25,10, 11, 14, 15, 69)col = c("#023FA5" ,"#5465AB" ,"#7D87B9" ,"#A1A6C8" ,"#BEC1D4")[5:1]
df <- data.frame(Rank = Rank, Completeness = Completeness, Number = Number)
df$Completeness = factor(df$Completeness , levels=c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))
df$Rank = factor(df$Rank , levels = c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Completeness)) + geom_bar(stat = 'identity', position = 'stack')+scale_fill_manual(values = col , limits= c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))+labs(x = "Binning method", y = "Number of bins", title = "CheckV results on the real human gut dataset")+coord_flip()+theme(legend.position="bottom",legend.title=element_text(size = 11),legend.text = element_text(size = 11),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 13,face = "bold"),axis.title.y = element_text(size = 13,face = "bold"),title = element_text(size = 14,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("fig3a.eps", width = 6.3, height = 5, device = cairo_ps)##############cow fecal 2b############
Rank = rep(c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'),each = 5)
Completeness = rep(c( "≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"),times = 5)
###Number needs to be 4*5 matrix##
Number = c(21 , 14 , 21 , 9 , 60,14 , 17 , 12 , 8 , 31,18, 14 , 16 , 14 , 36,3, 3 , 2 , 2 , 25,19,17,10,8,23)col = c("#023FA5" ,"#5465AB" ,"#7D87B9" ,"#A1A6C8" ,"#BEC1D4")[5:1]
df <- data.frame(Rank = Rank, Completeness = Completeness, Number = Number)
df$Completeness = factor(df$Completeness , levels=c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))
df$Rank = factor(df$Rank , levels = c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Completeness)) + geom_bar(stat = 'identity', position = 'stack')+scale_fill_manual(values = col , limits= c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))+labs(x = "Binning method", y = "Number of bins", title = "CheckV results on the real cow fecal dataset")+coord_flip()+theme(legend.position="bottom",legend.title=element_text(size = 11),legend.text = element_text(size = 11),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 13,face = "bold"),axis.title.y = element_text(size = 13,face = "bold"),title = element_text(size = 14,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("fig3b.eps", width = 6.3, height = 5, device = cairo_ps)##############wastewater 2c############
Rank = rep(c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'),each = 5)
Completeness = rep(c( "≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"),times = 5)
###Number needs to be 3*5 matrix##
Number = c(30 , 27 , 21 , 17 , 77,19, 20 , 11 , 11 , 28,14,16,14,15,32,2, 8 , 8 , 6 , 38,20,34,14,13,58)col = c("#023FA5" ,"#5465AB" ,"#7D87B9" ,"#A1A6C8" ,"#BEC1D4")[5:1]
df <- data.frame(Rank = Rank, Completeness = Completeness, Number = Number)
df$Completeness = factor(df$Completeness , levels=c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))
df$Rank = factor(df$Rank , levels = c('ViralCC' ,'bin3C' , 'vRhyme' , 'CoCoNet' , 'VAMB'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Completeness)) + geom_bar(stat = 'identity', position = 'stack')+scale_fill_manual(values = col , limits= c("≥ 50%", "≥ 60%", "≥ 70%", "≥ 80%" , "≥ 90%"))+labs(x = "Binning method", y = "Number of bins", title = "CheckV results on the real wastewater dataset")+coord_flip()+theme(legend.position="bottom",legend.title=element_text(size = 11),legend.text = element_text(size = 11),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 13,face = "bold"),axis.title.y = element_text(size = 13,face = "bold"),title = element_text(size = 14,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("fig3c.eps", width = 6.35, height = 5, device = cairo_ps)########Fraction of host by different number of viruses#########df<-data.frame(group=c('infected by one virus' , 'infected by two viruses', 'infected by three viruses'),value=c(25,35,45))
df$group = as.vector(df$group)ggplot(df,aes(x="",y=value,fill=group))+geom_bar(stat="identity")+coord_polar("y",start=1) + geom_text(aes(y=c(0,cumsum(value)[-length(value)]),label=percent(value/100)),size=5)+theme_minimal()+theme(axis.title=element_blank(),axis.ticks=element_blank(),axis.text = element_blank(),legend.title = element_blank())+scale_fill_manual(values=c("darkgreen","orange","deepskyblue"))##########Supplementary material###########
########Mock cow fecal dataset#######
Rank = rep(c('F-score' , 'ARI' , 'NMI' , 'Homogeneity') , each = 4)
Pipeline = rep(c( 'CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),times = 4)
Number = c(0.564, 0.763 , 0.936 , 0.936,0.455 ,0.719, 0.926 , 0.926,0.796 , 0.885 , 0.969 , 0.963,0.661 ,0.806, 0.940 , 1)col = c('#4FB0C6', "#4F86C6", "#527F76", '#CC9966')df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))
df$Rank = factor(df$Rank , levels = c('F-score' , 'ARI' , 'NMI', 'Homogeneity'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'dodge')+scale_fill_manual(values = col,limits= c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))+coord_cartesian(ylim = c(0.3,1))+labs(x = "Clustering metrics", y = "Scores", title = "The mock cow fecal dataset")+theme(legend.position="bottom",legend.title=element_blank(),legend.text = element_text(size = 12),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("sp1a.eps", width = 6, height = 5, device = cairo_ps)Rank = rep(c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 4)
Number = c(1 , 1 , 3 , 3,2,2,1, 3 , 5 , 0 ,0 , 8 )col = c("#8FBC94","#77AAAD","#6E7783")df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'stack')+coord_cartesian(ylim = c(0 , 9))+scale_y_discrete(limits = c(0 , 3 , 6 , 9))+scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+labs(x = "Binning method", y = "Number of viral bins", title = "The mock cow fecal dataset")+theme(legend.position="bottom",legend.title=element_blank(),legend.text = element_text(size = 12),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("sp1b.eps", width = 6, height = 5, device = cairo_ps)##########Supplementary material###########
########Mock wastewater fecal#######
Rank = rep(c('F-score' , 'ARI' , 'NMI' , 'Homogeneity') , each = 4)
Pipeline = rep(c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),times = 4)
Number = c(0.667,0.657,0.858,0.903,0.602 ,0.596,0.828,0.891,0.806 ,0.843, 0.898,0.937,0.687 ,0.746, 0.816,0.881)col = c('#4FB0C6', "#4F86C6", "#527F76", '#CC9966')df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))
df$Rank = factor(df$Rank , levels = c('F-score' , 'ARI' , 'NMI', 'Homogeneity'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'dodge')+scale_fill_manual(values = col,limits= c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))+coord_cartesian(ylim = c(0.1,0.97))+labs(x = "Clustering metrics", y = "Scores", title = "The mock wastewater dataset")+theme(legend.position="bottom",legend.title=element_blank(),legend.text = element_text(size = 12),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("sp1c.eps", width = 6, height = 5, device = cairo_ps)Rank = rep(c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 4)
Number = c( 5 , 3 , 1 , 1,2,2,1, 3 , 1 , 1 ,3 , 12 )col = c("#8FBC94","#77AAAD","#6E7783")df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('CoCoNet' , 'vRhyme', 'bin3C' , 'ViralCC'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'stack')+scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+labs(x = "Binning method", y = "Number of viral bins", title = "The mock wastewater dataset")+theme(legend.position="bottom",legend.title=element_blank(),legend.text = element_text(size = 12),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 12),axis.text.y = element_text(size = 12),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 16,face = "bold"),plot.title = element_text(hjust = 0.5))ggsave("sp1d.eps", width = 6, height = 5, device = cairo_ps)##########CheckM results#############
Rank = rep(c('MetaBAT2' , 'CoCoNet' , 'bin3C' , 'ViralCC'),each = 3)
Pipeline = rep(c('Moderately complete' , 'Substantially complete' , 'Near-complete'),times = 4)
Number = c(3 , 4 , 4 , 5 , 3 , 1 , 1, 3 , 1 , 2 ,2 , 12 )col = c("#8FBC94","#77AAAD","#6E7783")df <- data.frame(Rank = Rank, Pipeline = Pipeline, Number = Number)
df$Pipeline = factor(df$Pipeline , levels=c('Moderately complete' , 'Substantially complete' , 'Near-complete'))
df$Rank = factor(df$Rank , levels = c('MetaBAT2' , 'CoCoNet' , 'bin3C' , 'ViralCC'))ggplot(data = df, mapping = aes(x = Rank, y = Number, fill = Pipeline)) + geom_bar(stat = 'identity', position = 'stack')+scale_fill_manual(values = col,limits= c('Moderately complete' , 'Substantially complete' , 'Near-complete'))+labs(x = "Binning method", y = "Number of bins", title = "Mock wastewater dataset")+theme(legend.position="top",legend.title=element_blank(),legend.text = element_text(size = 11),panel.grid.major = element_blank(), #不显示网格线panel.grid.minor = element_blank(),axis.text.x = element_text(size = 11),axis.text.y = element_text(size = 11),axis.title.x = element_text(size = 14,face = "bold"),axis.title.y = element_text(size = 14,face = "bold"),title = element_text(size = 14,face = "bold"),plot.title = element_text(hjust = 0.5))#######Compute the length of viral contigs########
contig_info = read.csv('contig_viral_info_ww.csv' , sep = ',' , header = F)
min(contig_info[,3])
max(contig_info[,3])#######Chi-square testing############
tableR = matrix(c(72,96,264,36,38,90,21,24,49,38,42,80),nrow=3)
chisq.test(tableR,correct = F)removesmalls.pl
## removesmalls.pl
##!/usr/bin/perl
## perl removesmalls.pl 200 contigs.fasta > contigs-l200.fasta
use strict;
use warnings;my $minlen = shift or die "Error: `minlen` parameter not provided\n";
{local $/=">";while(<>) {chomp;next unless /\w/;s/>$//gs;my @chunk = split /\n/;my $header = shift @chunk;my $seqlen = length join "", @chunk;print ">$_" if($seqlen >= $minlen);}local $/="\n";
}相关文章:
快速、准确地检测和分类病毒序列分析工具 ViralCC的介绍和详细使用方法, 附带应用脚本
介绍 viralcc是一个基因组病毒分析工具,可以用于快速、准确地检测和分类病毒序列。 github:dyxstat/ViralCC: ViralCC: leveraging metagenomic proximity-ligation to retrieve complete viral genomes (github.com) Instruction of reproducing resul…...

DNs服务学习笔记
DNS:域名系统(英文:Domain Name System)是一个域名系统,是万维网上作为域名和IP地址相互映射的一个分布式数据库,能够使用户更方便的访问互联网,而不用去记住能够被机器直接读取的IP数串。类似于生活中的11…...

获取线程池中任务执行数量
获取线程池中任务执行数量 通过线程池进行任务处理,有时我们需要知道线程池中任务的执行状态。通过ThreadPoolExecutor的相关API实时获取线程数量,排队任务数量,执行完成线程数量等信息。 实例 private static ExecutorService es new Thr…...

RK3566 Android 11平台上适配YT8512C 100M PHY
RK3566代码之前适配的1000M IC RTL8211F , 现在需要在之前的基础上修改PHY IC 为裕泰的YT8512C ----------------------------------------------------------------------//将1000M 的配置关掉,改为100M 配置,查看RK3566 资料关于以太网的配置即可知道如何修改 #if…...
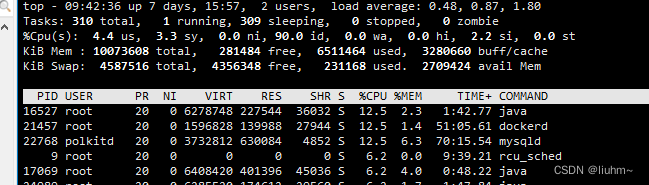
docker 部署haproxy cpu占用特别高
在部署mysql 主主高可用时,使用haproxy进行负载,在服务部使用的情况下发现服务器cpu占比高,负载也高,因此急需解决这个问题。 1.解决前现状 1.1 部署配置文件 cat > haproxy.cfg << EOF globalmaxconn 4000nbthrea…...
Oracle导出CSV文件
利用spool spool基本格式: spool 路径文件名 select col1||,||col2||,||col3||,||col4 from tablename; spool off spool常用的设置: set colsep ; //域输出分隔符 set echo off; //显示start启动的脚本中的每个sql命令,缺…...

图像分割实战-系列教程12:deeplab系列算法概述
🍁🍁🍁图像分割实战-系列教程 总目录 有任何问题欢迎在下面留言 本篇文章的代码运行界面均在Pycharm中进行 本篇文章配套的代码资源已经上传 1、deeplab概述 图像分割中的传统做法:为了增大感受野,通常都会选择pooling…...

数据库02-07 存储
计算机存储系统: 02.磁道存储...
WPF 入门教程DispatcherTimer计时器
https://www.zhihu.com/tardis/bd/art/430630047?source_id1001 在 WinForms 中,有一个名为 Timer 的控件,它可以在给定的时间间隔内重复执行一个操作。WPF 也有这种可能性,但我们有DispatcherTimer控件,而不是不可见的控件。它几…...
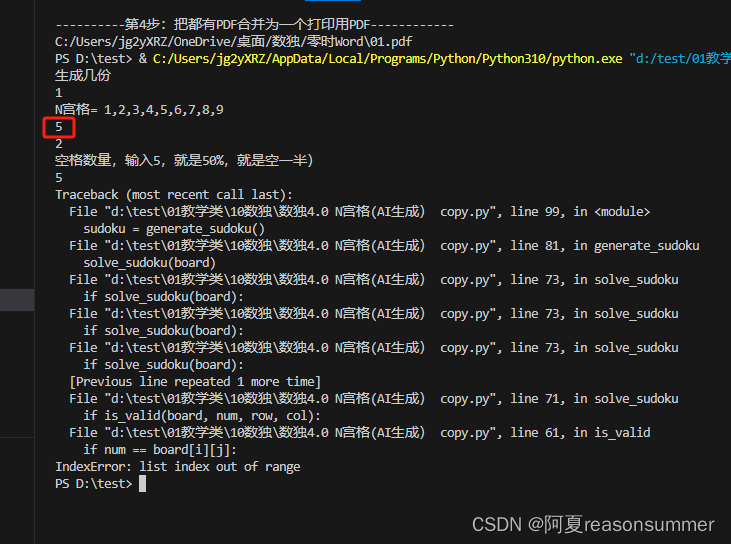
【教学类-43-04】20231229 N宫格数独4.0(n=2,4,6,8) (ChatGPT AI对话大师生成 回溯算法)
作品展示: 背景需求: 幼儿表示自己适合做5宫格 第一次AI生成九宫格数独python代码 【教学类-43-03】20231229 N宫格数独3.0(n1、2、3、4、6、8、9) (ChatGPT AI对话大师生成)-CSDN博客文章浏览阅读162次&…...

WPF美化ItemsControl1:不同颜色间隔
首先我们有的是一个绑定好数据的ItemsControl <ItemsControl ItemsSource"{Binding Starts}"> </ItemsControl> 运行后呢是朴素的将数据竖着排列 如果想要数据之间有间距,可以使用数据模板,将数据放到TextBlock中显示࿰…...
查看进程对应的路径查看端口号对应的进程ubuntu 安装ssh共享WiFi设置MyBatis 使用map类型作为参数,复杂查询(导出数据)
Linux 查询当前进程所在的路径 top 命令查询相应的进程号pid ps -ef |grep 进程名 lsof -I:端口号 netstat -anp|grep 端口号 cd /proc/进程id cwd 进程运行目录 exe 执行程序的绝对路径 cmdline 程序运行时输入的命令行命令 environ 记录了进程运行时的环境变量 fd 目录下是进…...

医院信息系统集成平台—安全保障体系
隐私保护措施 隐私保护及信息安全是医院信息平台所要重点解决的问题,应从患者同意,匿名化服务,依据病种、角色等多维度授权,关键信息(字段级、记录级、文件级)加密存储等方面展开。电子病历等医疗数据进行调阅时,包括强身份认证需求、角色授权需求、责任认…...

【信息论与编码】习题-填空题
目录 填空题1.克劳夫特不等式是判断( )的充要条件。2.无失真信源编码的中心任务是编码后的信息率压缩接近到()限失真压缩中心任务是在给定的失真度条件下,信息率压缩接近到( )。3.常用的检纠错方…...
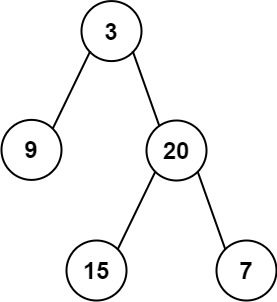
二叉树的层序遍历经典问题(算法村第六关白银挑战)
基本的层序遍历与变换 二叉树的层序遍历 102. 二叉树的层序遍历 - 力扣(LeetCode) 给你二叉树的根节点 root ,返回其节点值的 层序遍历 。 (即逐层地,从左到右访问所有节点)。 示例 1: 输入…...

信息学奥赛一本通:装箱问题
题目链接:http://ybt.ssoier.cn:8088/problem_show.php?pid1917 题目 1917:【01NOIP普及组】装箱问题 时间限制: 1000 ms 内存限制: 65536 KB 提交数: 4117 通过数: 2443 【题目描述】 有一个箱子容量为V�(正整数,…...
ReactNative 常见问题及处理办法(加固混淆)
ReactNative 常见问题及处理办法(加固混淆) 文章目录 摘要引言正文ScrollView内无法滑动RN热更新中的文件引用问题RN中获取高度的技巧RN强制横屏UI适配问题低版本RN(0.63以下)适配iOS14图片无法显示问题RN清理缓存RN navigation参…...

算法基础之合并果子
合并果子 核心思想: 贪心 Huffman树(算法): 每次将两个最小的堆合并 然后不断向上合并 #include<iostream>#include<algorithm>#include<queue> //用小根堆实现找最小堆using namespace std;int main(){int n;cin>>n;priority_queue&l…...

CSS 使用技巧
CSS 使用技巧 引入苹方字体 苹方提供了六个字重,font-family 定义如下:苹方-简 常规体font-family: PingFangSC-Regular, sans-serif;苹方-简 极细体font-family: PingFangSC-Ultralight, sans-serif;苹方-简 细体font-family: PingFangSC-Light, sans…...

typescript,eslint,prettier的引入
typescript 首先用npm安装typescript,cnpm i typescript 然后再tsc --init生成tsconfig.json配置文件,这个文件在package.json同级目录下 最后在tsconfig.json添加includes配置项,在该配置项中的目录下,所有的d.ts中的类型可以在…...

Data Storage and Computation
Data Storage and Computation 数据存储与计算假设一张表有 3 个字段:id BIGINT(8 字节 / 条) name VARCHAR(20)(实际平均 10 字节 / 条) age TINYINT(1 字节 / 条)单行实际数据占用࿱…...

告别图形界面:在Linux终端中高效管理百度网盘文件的完整指南
1. 为什么需要命令行管理百度网盘? 很多开发者都遇到过这样的场景:远程连接到Linux服务器时,需要快速上传日志文件到网盘,或者从网盘下载数据集到服务器。传统做法是先把文件下载到本地电脑,再用SFTP工具上传到服务器—…...

【2026实测】直击算法底层逻辑:论文AI率太高?5款工具与3大手改技巧盘点
最近不少学弟学妹在后台跟我倒苦水,说查重率好不容易低了,结果AI率越改越高。眼看临近DDL,生怕又因为这个耽误答辩。 作为已经摸爬滚打出来的老学长,今天我就根据我总结出来的经验,从检测系统的底层逻辑开始讲起&…...

前端开发者福音:用Vue.js开发的Beekeeper Studio,如何让SQL开发体验更‘现代’?
Vue.js与SQL工具的现代融合:Beekeeper Studio如何重塑开发者体验 当SQL开发者第一次打开Beekeeper Studio时,那种流畅的界面过渡和即时的语法补全反馈会让人误以为在使用一个现代Web应用——这恰恰是Vue.js赋予桌面应用的魔力。作为一款基于Electron和Vu…...

【LangChain】 入门:从分步调用到链式编程
LangChain 入门:从分步调用到链式编程本文基于一段翻译助手的示例代码,讲解 LangChain 的核心概念、输出解析器的作用,以及普通写法与链式写法的对比。一、LangChain 是什么? 名字拆解缩写含义LangLanguage(语言&#…...

GPU硬件操作强度与LLM推理效率优化实践
1. 硬件操作强度(HOI)与LLM推理效率的深度解析在GPU加速的大型语言模型推理场景中,我们常常遇到一个看似矛盾的现象:计算单元利用率不足的同时,显存带宽却成为瓶颈。这种现象的根源在于硬件操作强度(Hardwa…...

Vibe Coding:打造沉浸式编程学习环境,从环境到心流的高效开发实践
1. 项目概述:从“Vibe Coding”到沉浸式编程学习 最近在开发者社区里,一个名为“VibecodingCurriculum”的项目引起了我的注意。这个由 hashed 团队在 vibedojo 下维护的仓库,名字本身就很有意思——“Vibe Coding”,直译过来是“…...
)
别再搞混了!Web地图开发必懂的EPSG:4326和EPSG:3857(附JavaScript转换代码)
Web地图开发中的坐标系解密:从原理到实战 第一次在Leaflet地图上叠加GPS轨迹数据时,我盯着那个偏离了三条街的路径百思不得其解——经纬度坐标明明正确,为什么显示位置完全不对?这个困扰无数Web开发者的经典问题,根源在…...

为AI智能体构建长期记忆系统:零配置集成与四通道混合检索实践
1. 项目概述:为AI智能体装上“长期记忆”在AI智能体(Agent)的开发与使用中,一个长期存在的痛点就是“健忘症”。无论是基于OpenAI API还是本地部署的大模型,标准的对话模式都是无状态的——每次交互对于模型来说都是一…...

使用remote2mac实现Windows远程开发macOS:VSCode SSH配置与优化指南
1. 项目概述与核心价值最近在折腾远程开发环境,特别是需要在不同操作系统间无缝切换时,遇到了一个挺典型的痛点:手头的主力开发机是Windows,但项目部署和测试环境往往是macOS或Linux服务器。传统的远程桌面方案要么延迟高得没法写…...