indel_snp_ssr_primer
indel标记使用
1.得到vcf文件
2.提取指定区域vcf文件并压缩构建索引
bcftools view -r <CHROM>:<START>-<END> input.vcf -o output.vcf
bgzip -c all.filtered.indel.vcf > all.filtered.indel.vcf.gz
tabix -p vcf all.filtered.indel.vcf.gz
3.准备参考基因组文件
4.配置perl环境,注意区分系统自带的perl和conda安装的perl,在安装perl模块时指定使用的是哪个perl,否则会出现安装了模块但无法识别
5.primer3 不同版本的default_settings.txt在不同目录,注意查看位置并在primer_indel.pl脚本中修改路径,如primer3(v2.6.1)在 ~/public_software/primer/primer3-main/settings_files/p3_th_settings.txt
6.最最重要dangle.dh文件,需要在p3_th_settings.txt中修改路径
示例命令
perl primer_indel.pl -fa duo_shao/Zea_mays.Zm-B73-REFERENCE-NAM-5.0.dna.toplevel.fa -vcf duo_shao/chr1_44105421_45153997.vcf -od duo_shao/chr1_44105421_45153997
第一部分:脚本头部和初始化
2. 模块加载
#use strict;
use Cwd qw(abs_path getcwd);
use Getopt::Long;
use Data::Dumper;
use FindBin qw($Bin $Script);
use File::Basename qw(basename dirname);
use Config::General;use File::Path qw(make_path remove_tree);
use File::Copy;
use File::Spec;
my $BEGIN_TIME=time();
my $version="1.0";
use Bio::SeqIO;
use Cwd qw(abs_path getcwd);:加载Cwd模块,用于获取当前工作目录和绝对路径。use Getopt::Long;:用于处理命令行参数。use Data::Dumper;:用于调试,可以打印复杂数据结构。use FindBin qw($Bin $Script);:获取脚本所在的目录和脚本名称。use File::Basename qw(basename dirname);:用于处理文件名和路径。use Config::General;:用于解析配置文件。use File::Path qw(make_path remove_tree);:用于创建和删除目录。use File::Copy;:用于文件复制操作。use File::Spec;:用于处理文件路径。use Bio::SeqIO;:用于读取和处理生物序列文件(如 FASTA 格式)。
3. 命令行参数解析
my ($vcf,$flank,$fa,$epcrfa,$od,$outprefix,$PRIMER_NUM_RETURN,$PRIMER_OPT_SIZE,$PRIMER_MIN_SIZE,$PRIMER_MAX_SIZE,$PRIMER_PRODUCT_SIZE_RANGE,$PRIMER_MAX_NS_ACCEPTED);GetOptions("help|?" =>\&USAGE,"fa=s"=>\$fa,"vcf=s"=>\$vcf,"flank=s"=>\$flank,"PRIMER_MIN_SIZE=s"=>\$PRIMER_MIN_SIZE,"PRIMER_OPT_SIZE=s"=>\$PRIMER_OPT_SIZE,"PRIMER_MAX_SIZE=s"=>\$PRIMER_MAX_SIZE,"PRIMER_NUM_RETURN=s"=>\$PRIMER_NUM_RETURN,"PRIMER_PRODUCT_SIZE_RANGE=s"=>\$PRIMER_PRODUCT_SIZE_RANGE,"epcrfa=s"=>\$epcrfa,"od=s"=>\$od,"prefix=s"=>\$outprefix,
) or &USAGE;
&USAGE unless($fa and $vcf);
GetOptions:解析命令行参数,将参数值赋给相应的变量。-fa:输入的参考基因组文件(FASTA 格式)。-vcf:输入的 VCF 文件。-flank:indel 两侧的侧翼序列长度,默认为 200。-PRIMER_MIN_SIZE、-PRIMER_OPT_SIZE、-PRIMER_MAX_SIZE:引物的最小、最优和最大长度。-PRIMER_NUM_RETURN:返回的引物对数量。-PRIMER_PRODUCT_SIZE_RANGE:引物扩增产物的长度范围。-epcrfa:用于电子 PCR 的参考基因组文件。-od:输出目录,默认为当前工作目录。-prefix:输出文件的前缀,默认为indel。
&USAGE:如果用户未提供必要的参数(-fa和-vcf),则调用USAGE子程序,显示帮助信息并退出。
4. 帮助信息
sub USAGE {my $usage=<<"USAGE";
Program: $0
Version: $version
Contact: huangls
Discription:Usage:Options:-fa <file> Input fa file, forced-vcf <file> Input indel vcf file, forced-flank 200 cut 200 ; -PRIMER_MIN_SIZE 18 Minimum acceptable length of a primer. Must be greater than 0 and less than or equal to PRIMER_MAX_SIZE-PRIMER_OPT_SIZE 20 Optimum length (in bases) of a primer. Primer3 will attempt to pick primers close to this length.-PRIMER_MAX_SIZE 23 Maximum acceptable length (in bases) of a primer. Currently this parameter cannot be larger than 35. This limit is governed by maximum oligo size for which primer3's melting-temperature is valid.-PRIMER_PRODUCT_SIZE_RANGE 100-1000 The associated values specify the lengths of the product that the userwants the primers to create, and is a space separated list of elements of the form; -PRIMER_NUM_RETURN 1 Total num primer to search -epcrfa run E-PCR to filter multi mapped primers; -od <str> Key of output dir,default cwd;-prefix <str> defoult demo;-h HelpUSAGEprint $usage;exit 1;
}
USAGE子程序:定义了脚本的使用说明,包括所有支持的命令行参数及其说明。
总结
这一部分主要完成了以下任务:
- 脚本头部信息和模块加载。
- 命令行参数的解析。
- 提供帮助信息。
第二部分:默认参数设置和全局变量初始化
1. 设置默认参数值
$PRIMER_MIN_SIZE ||= 18;
$PRIMER_OPT_SIZE ||= 20;
$PRIMER_MAX_SIZE ||= 23;
$PRIMER_NUM_RETURN ||= 1;
$PRIMER_MAX_NS_ACCEPTED ||= 1;
$PRIMER_PRODUCT_SIZE_RANGE ||= "100-1000";
$od ||= getcwd;
$od = abs_path($od);
unless (-d $od) { mkdir $od; }
$outprefix ||= "indel";
- 作用:为未在命令行中指定的参数设置默认值。
PRIMER_MIN_SIZE:引物的最小长度,默认为 18。PRIMER_OPT_SIZE:引物的最优长度,默认为 20。PRIMER_MAX_SIZE:引物的最大长度,默认为 23。PRIMER_NUM_RETURN:返回的引物对数量,默认为 1。PRIMER_MAX_NS_ACCEPTED:引物中允许的最大N(未知碱基)数量,默认为 1。PRIMER_PRODUCT_SIZE_RANGE:引物扩增产物的长度范围,默认为100-1000。od:输出目录,默认为当前工作目录。outprefix:输出文件的前缀,默认为indel。
getcwd和abs_path:获取当前工作目录的绝对路径,并确保输出目录存在。
2. 初始化全局变量
my %indel_length_range = ('mi' => 2, 'ma' => 5);
my %ref = ();
my %ref_len = ();
%indel_length_range:定义了 indel 长度的范围,mi表示最小长度,ma表示最大长度。%ref和%ref_len:用于存储参考基因组序列及其长度。
3. 读取参考基因组文件
my $in = Bio::SeqIO->new(-file => "$fa", -format => 'Fasta');
while (my $seq = $in->next_seq()) {$ref{$seq->id} = $seq;$ref_len{$seq->id} = $seq->length;
}
$in->close();
Bio::SeqIO:用于读取 FASTA 格式的参考基因组文件。$seq->id:获取序列的 ID(通常是染色体编号)。$seq->length:获取序列的长度。- 作用:将参考基因组序列及其长度存储到
%ref和%ref_len哈希表中,方便后续使用。
总结
这一部分主要完成了以下任务:
- 为未指定的参数设置默认值。
- 初始化全局变量,用于存储参考基因组序列及其长度。
- 读取参考基因组文件,并将其存储到哈希表中。
第三部分:VCF 文件的读取和处理
1. 处理 VCF 文件路径
$fa = abs_path($fa);
$epcrfa ||= $fa;
$epcrfa = abs_path($epcrfa);
- 作用:
- 将输入的参考基因组文件路径(
$fa)转换为绝对路径。 - 如果未指定用于电子 PCR 的参考基因组文件(
$epcrfa),则默认使用$fa。 - 确保
$epcrfa也是绝对路径。
- 将输入的参考基因组文件路径(
2. 打开 VCF 文件
if ($vcf =~ /gz$/) {open IN, "gzip -dc $vcf|" or die "$! $vcf";
} else {open IN, "$vcf" or die "$! $vcf";
}
- 作用:
- 检查 VCF 文件是否为 gzip 压缩文件(通过文件扩展名
.gz判断)。 - 如果是压缩文件,使用
gzip -dc命令解压并读取内容。 - 如果不是压缩文件,直接打开文件。
- 如果文件无法打开,程序将终止并报错。
- 检查 VCF 文件是否为 gzip 压缩文件(通过文件扩展名
3. 初始化变量
$flank ||= 200;
my %ssr_pos = ();
my %SSR = ();
my %genotype = ();unless (-d "$od/split") { mkdir "$od/split"; }
open OUT, ">$od/split/p_in_0.txt" or die "$!";
open SH, ">$od/primer3.sh" or die "$!";
$flank:设置 indel 两侧的侧翼序列长度,默认为 200。%ssr_pos和%SSR:用于存储 SSR(简单序列重复)的位置信息和 SSR 序列。%genotype:用于存储每个 indel 的基因型信息。mkdir "$od/split":创建一个目录用于存放 Primer3 的输入文件。open OUT和open SH:分别打开两个文件句柄,用于写入 Primer3 的输入文件和 Shell 脚本。
4. 读取 VCF 文件内容
while (my $line = <IN>) {chomp $line;next if $line =~ /^##/;# next if $line =~ /Scaffold/i;my @tmp = split(/\t/, $line);if ($line =~ /^#C/) {@{$genotype{head}} = @tmp[7 .. $#tmp];next;}my ($ref_len, $alt_len) = (length $tmp[3], length $tmp[4]);my $indel_len = abs($ref_len - $alt_len);my $indel_len_s = $alt_len - $ref_len;my $ID = "$tmp[0]_$tmp[1]_$indel_len";$ssr_pos{$ID} = "$tmp[0]\t$tmp[1]\t$tmp[3]\t$tmp[4]\t$indel_len_s";$tmp[7] =~ s/^.*ANNOVAR_DATE=[^;]+;//;@{$genotype{$ID}} = @tmp[7 .. $#tmp];my $seq = "";my $indel_len_target = $indel_len;if ($alt_len > 1) {$seq = &get_target_fa($tmp[0], $tmp[1] - $flank, $tmp[1] + $flank, $flank, $tmp[4]);$indel_len_target = 1;} else {$seq = &get_target_fa($tmp[0], $tmp[1] - $flank, $tmp[1] + $flank + $indel_len, $flank);}
- 作用:
- 逐行读取 VCF 文件内容。
- 跳过以
##开头的注释行。 - 如果是列头行(以
#C开头),提取样本信息并存储到%genotype{head}中。 - 对于每个 indel 变异:
- 计算参考碱基(
REF)和变异碱基(ALT)的长度。 - 计算 indel 的长度(
indel_len)和符号(indel_len_s)。 - 构造一个唯一的 ID(
$ID),格式为染色体编号_位置_indel长度。 - 将 indel 的位置信息存储到
%ssr_pos中。 - 提取该 indel 的基因型信息并存储到
%genotype中。 - 调用
get_target_fa函数获取 indel 两侧的侧翼序列($seq)。 - 如果
ALT是插入变异(长度 > 1),将目标长度设置为 1;否则,目标长度为 indel 的实际长度。
- 计算参考碱基(
5. 检测 SSR 并生成 Primer3 输入文件
if ($seq) {my ($is_have_ssr, $ssr) = &has_ssr($ID, $seq);if ($is_have_ssr) {$SSR{$ID} = join(",", @{$ssr});} else {$SSR{$ID} = "--";}print OUT "SEQUENCE_ID=$ID\n";print OUT "SEQUENCE_TEMPLATE=$seq\n";printf OUT ("SEQUENCE_TARGET=%d,%d\n", $flank + 1, $indel_len_target);print OUT "PRIMER_TASK=pick_detection_primers\n";print OUT "PRIMER_PICK_LEFT_PRIMER=1\n";print OUT "PRIMER_PICK_RIGHT_PRIMER=1\n";print OUT "PRIMER_NUM_RETURN=$PRIMER_NUM_RETURN\n";print OUT "PRIMER_MAX_END_STABILITY=250\n";print OUT "PRIMER_MIN_SIZE=$PRIMER_MIN_SIZE\n";print OUT "PRIMER_OPT_SIZE=$PRIMER_OPT_SIZE\n";print OUT "PRIMER_MAX_SIZE=$PRIMER_MAX_SIZE\n";print OUT "PRIMER_PRODUCT_OPT_SIZE=125\n";print OUT "PRIMER_OPT_GC_PERCENT=50\n";print OUT "PRIMER_MIN_TM=50\n";print OUT "PRIMER_OPT_TM=55\n";print OUT "PRIMER_MAX_TM=65\n";print OUT "PRIMER_MAX_NS_ACCEPTED=1\n";print OUT "PRIMER_FIRST_BASE_INDEX=1\n";print OUT "PRIMER_MIN_GC=40.0\nPRIMER_MAX_GC=60.0\n";print OUT "PRIMER_PRODUCT_SIZE_RANGE=$PRIMER_PRODUCT_SIZE_RANGE\n";printf OUT ("SEQUENCE_INTERNAL_EXCLUDED_REGION=%d,%d\n", $flank + 1, $indel_len_target);print OUT "=\n";$n++;if ($n > 100) {$n = 0;$num++;close(OUT);open OUT, ">$od/split/p_in_$num.txt" or die "$!";print SH "primer3_core -p3_settings_file=/share/work/biosoft/primer3/latest/default_settings.txt -default_version=1 -output=$od/split/${num}primers.txt $od/split/p_in_$num.txt\n";}
}
- 作用:
- 如果成功获取了侧翼序列(
$seq),则调用has_ssr函数检查该序列是否包含 SSR。 - 如果存在 SSR,将 SSR 信息存储到
%SSR中;否则,存储--。 - 生成 Primer3 的输入文件内容,包括序列 ID、模板序列、目标区域等信息。
- 每处理 100 个 indel,将 Primer3 输入文件分批保存到不同的文件中,并生成对应的 Shell 命令行。
- 这样可以避免单个 Primer3 输入文件过大,提高运行效率。
- 如果成功获取了侧翼序列(
总结
这一部分主要完成了以下任务:
- 处理 VCF 文件路径,确保文件可以正确读取。
- 初始化相关变量,用于存储 SSR 和基因型信息。
- 逐行读取 VCF 文件,提取 indel 信息,并获取 indel 两侧的侧翼序列。
- 检测 SSR,并生成 Primer3 的输入文件。
- 将 Primer3 输入文件分批保存,生成对应的 Shell 命令行。
第四部分:运行 Primer3 和电子 PCR(e-PCR)
1. 关闭文件句柄并生成 Primer3 脚本
close(OUT);
close(IN);
close(SH);#system("sh $od/primer3.sh");
system("parallel -j 10 <$od/primer3.sh");
system("cat $od/split/*primers.txt >$od/$outprefix.primers");
- 作用:
- 关闭所有打开的文件句柄。
- 使用
parallel命令并行运行 Primer3,提高效率。-j 10表示同时运行 10 个任务。 - 将所有分批生成的 Primer3 输出文件合并为一个文件,命名为
$outprefix.primers。
2. 解析 Primer3 输出文件
$/ = "=\n";
my %Pseq = ();open IN, "$od/$outprefix.primers" or die "$!";
open OUT, ">$od/$outprefix.primers.xls" or die "$!";
print OUT "#SEQUENCE_ID\tPRIMER_LEFT_SEQUENCE\tPRIMER_RIGHT_SEQUENCE\tPRIMER_PAIR_PRODUCT_SIZE\tPRIMER_LEFT_TM\tPRIMER_RIGHT_TM\tPRIMER_LEFT_GC_PERCENT\tPRIMER_RIGHT_GC_PERCENT\tSSR\tSSR_TYPE\n";
while (my $line = <IN>) {chomp $line;my @field = split(/\n/, $line);my %primers = &get_hash(@field);next if !defined($primers{"PRIMER_PAIR_NUM_RETURNED"}) or $primers{"PRIMER_PAIR_NUM_RETURNED"} == 0;if ($primers{"PRIMER_PAIR_NUM_RETURNED"} == 1) {$Pseq{"$primers{SEQUENCE_ID}"} = [$primers{"PRIMER_LEFT_0_SEQUENCE"}, $primers{"PRIMER_RIGHT_0_SEQUENCE"}];print OUT join("\t", ($primers{"SEQUENCE_ID"}, $primers{"PRIMER_LEFT_0_SEQUENCE"}, $primers{"PRIMER_RIGHT_0_SEQUENCE"}, $primers{"PRIMER_PAIR_0_PRODUCT_SIZE"}, $primers{"PRIMER_LEFT_0_TM"}, $primers{"PRIMER_RIGHT_0_TM"}, $primers{"PRIMER_LEFT_0_GC_PERCENT"}, $primers{"PRIMER_RIGHT_0_GC_PERCENT"}, $SSR{$primers{"SEQUENCE_ID"}})) . "\n";} else {for my $i (0 .. ($primers{"PRIMER_PAIR_NUM_RETURNED"} - 1)) {$Pseq{"$primers{SEQUENCE_ID}.$i"} = [$primers{"PRIMER_LEFT_${i}_SEQUENCE"}, $primers{"PRIMER_RIGHT_${i}_SEQUENCE"}];print OUT join("\t", ("$primers{SEQUENCE_ID}.$i", $primers{"PRIMER_LEFT_${i}_SEQUENCE"}, $primers{"PRIMER_RIGHT_${i}_SEQUENCE"}, $primers{"PRIMER_PAIR_${i}_PRODUCT_SIZE"}, $primers{"PRIMER_LEFT_${i}_TM"}, $primers{"PRIMER_RIGHT_${i}_TM"}, $primers{"PRIMER_LEFT_${i}_GC_PERCENT"}, $primers{"PRIMER_RIGHT_${i}_GC_PERCENT"}, $SSR{$primers{"SEQUENCE_ID"}})) . "\n";}}
}
$/ = "\n";
close(OUT);
- 作用:
- 设置输入记录分隔符
$/为=\n,以便正确解析 Primer3 的输出文件。 - 打开 Primer3 输出文件和结果文件。
- 遍历 Primer3 输出文件的每一部分(以
=分隔),解析引物设计结果。 - 调用
get_hash函数将 Primer3 输出转换为哈希表。 - 如果某个序列没有设计出引物,则跳过。
- 如果设计出了一对引物,将其存储到
%Pseq中,并将相关信息写入结果文件。 - 如果设计出了多对引物,将每对引物的信息分别存储和写入结果文件。
- 最后,将输入记录分隔符恢复为默认值
\n。
- 设置输入记录分隔符
3. 运行电子 PCR(e-PCR)
print "e-PCR -w9 -f 1 -m100 $od/$outprefix.primers.xls D=$PRIMER_PRODUCT_SIZE_RANGE $epcrfa N=0 G=0 T=3 > $od/$outprefix.epcr.out\n";
system("e-PCR -w9 -f 1 -m100 $od/$outprefix.primers.xls D=$PRIMER_PRODUCT_SIZE_RANGE $epcrfa N=0 G=0 T=3 > $od/$outprefix.epcr.out");
- 作用:
- 构造 e-PCR 命令行,运行 e-PCR 检查引物的特异性。
- 参数说明:
-w9:允许最多 9 个错配。-f 1:只考虑正链。-m100:最大产物长度为 100。D=$PRIMER_PRODUCT_SIZE_RANGE:引物产物长度范围。$epcrfa:用于 e-PCR 的参考基因组文件。N=0:不允许未定义的核苷酸(N)。G=0:不允许间隙。T=3:使用 3 个核苷酸的窗口进行匹配。
- 将 e-PCR 的输出保存到
$od/$outprefix.epcr.out文件中。
4. 解析 e-PCR 输出文件
open IN, "$od/$outprefix.epcr.out" or die "$!";
open OUT, ">$od/$outprefix.primers.result.xls" or die "$!";
my %P = ();
while (my $line = <IN>) {chomp $line;my @tmp = split(/\t/, $line);next if $tmp[6] + $tmp[7] > 1;$tmp[5] =~ s#/.*$##;$P{$tmp[1]}{"$tmp[1]_$tmp[3]_$tmp[4]"} = [$tmp[1], $ssr_pos{$tmp[1]}, $Pseq{$tmp[1]}->[0], $Pseq{$tmp[1]}->[1], $tmp[5], @tmp[6 .. $#tmp], @{$genotype{$tmp[1]}}];
}
close(IN);
print OUT "#PRIMER_ID\tCHROM\tPOS\tREF\tALT\tINDEL_LENGTH\tPRIMER_LEFT_SEQUENCE\tPRIMER_RIGHT_SEQUENCE\tPRIMER_PAIR_PRODUCT_SIZE\tMISM\tGAPS\tPRIMER_LEFT_TM\tPRIMER_RIGHT_TM\tPRIMER_LEFT_GC_PERCENT\tPRIMER_RIGHT_GC_PERCENT\tHAS_SSR\t" . join("\t", @{$genotype{head}}) . "\n";for my $id (sort keys %P) {my @ps = (keys %{$P{$id}});if (@ps == 1) {print OUT join("\t", @{$P{$id}{$ps[0]}}) . "\n";}
}
close(OUT);
- 作用:
- 打开 e-PCR 输出文件和最终结果文件。
- 遍历 e-PCR 输出文件的每一行,解析引物的匹配结果。
- 跳过匹配到多个位置的引物(
$tmp[6] + $tmp[7] > 1)。 - 提取引物的相关信息,并存储到
%P哈希表中。 - 将最终结果写入
$od/$outprefix.primers.result.xls文件,包括引物 ID、染色体位置、引物序列、产物长度、错配数、间隙数、引物退火温度、GC 含量、是否含有 SSR 以及基因型信息。
总结
这一部分主要完成了以下任务:
- 关闭文件句柄并生成 Primer3 脚本。
- 解析 Primer3 输出文件,提取引物设计结果。
- 运行电子 PCR(e-PCR),检查引物的特异性。
- 解析 e-PCR 输出文件,生成最终的引物结果文件。
好的,接下来我们分析脚本的第五部分:生成 README 文件和清理临时文件。
第五部分:生成 README 文件和清理临时文件
1. 生成 README 文件
open OUT, ">$od/readme.txt" or die "$!";
my $readme = <<"README";
*primers.result.xls
PRIMER_ID 引物ID (命名规则:indel所在来源序列_indel所在位置start_indel所在位置长度)
CHROM 所在染色体
POS 所在染色体位置
REF 参考基因组碱基
ALT 变异碱基
INDEL_LENGTH indel长度(负数表示缺失,正数表示插入)
PRIMER_LEFT_SEQUENCE-- 左引物
PRIMER_RIGHT_SEQUENCE-- 右引物
PRIMER_PAIR_PRODUCT_SIZE-- 引物产物长度
MISM-- Total number of mismatches for two primers(ePCR)
GAPS-- Total number of gaps for two primers(ePCR)
PRIMER_LEFT_TM-- 退火温度
PRIMER_RIGHT_TM-- 退火温度
PRIMER_LEFT_GC_PERCENT-- GC含量
PRIMER_RIGHT_GC_PERCENT-- GC含量
HAS_SSR-- indel周围是否含有SSR -- 表示不含有SSR
INFO vcf注释信息 参照变异检测说明FORMAT vcf注释信息 参照变异检测说明 http://www.omicsclass.com/article/6 AD,"Allelic depths for the ref and alt alleles in the order listed"DP,"Approximate read depth (reads with MQ=255 or with bad mates are filtered)"GQ,"Genotype Quality"GT,"Genotype" 0表示和参考基因型相同(REF),1表示变异基因型(ALT)。PL,"Normalized, Phred-scaled likelihoods for genotypes as defined in the VCF specification"
样品基因型 信息,用":"隔开与FORMAT列对应方法:
1.提取indel左右各$flank bp序列,用primer3设计引物[2]
2.用Epcr ,在参考基因组上电子PCR,去除有多处比对的引物,保证引物扩增的特异性[3]
3.检测indel附近是否有SSR[1][1] MISA:Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.).
[2] Primer3: Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3 - new capabilities and interfaces. Nucleic Acids Research 40(15):e115
[3] Schuler, G. D. (1997). Sequence Mapping by Electronic PCR. Genome Research, 7(5), 541–550.READMEprint OUT $readme;
close(OUT);
- 作用:
- 打开一个文件句柄,准备写入 README 文件。
- 构造一个字符串
$readme,包含对输出文件格式和内容的详细说明。 - 写入 README 文件,解释每个字段的含义,包括引物 ID、染色体位置、引物序列、产物长度、错配数、间隙数、退火温度、GC 含量、SSR 信息以及 VCF 注释信息。
- 说明了脚本运行的方法和引用的工具(如 Primer3 和 e-PCR)。
- 关闭 README 文件。
2. 清理临时文件
print "head -1 $od/$outprefix.primers.result.xls >$od/head && cat $od/$outprefix.primers.result.xls|grep -v '#' |sort -k2,2 -k3,3n >$od/$outprefix.primers.result.xls.sorted && cat $od/head $od/$outprefix.primers.result.xls.sorted >$od/$outprefix.primers.result.xls && rm $od/head $od/$outprefix.primers.result.xls.sorted\n";
system "head -1 $od/$outprefix.primers.result.xls >$od/head && cat $od/$outprefix.primers.result.xls|grep -v '#' |sort -k2,2 -k3,3n >$od/$outprefix.primers.result.xls.sorted && cat $od/head $od/$outprefix.primers.result.xls.sorted >$od/$outprefix.primers.result.xls && rm $od/head $od/$outprefix.primers.result.xls.sorted";
- 作用:
- 使用
head命令提取结果文件的第一行(表头)。 - 使用
grep命令过滤掉以#开头的注释行。 - 使用
sort命令按染色体编号和位置对结果进行排序。 - 将排序后的结果重新组合为一个完整的文件。
- 删除临时文件(如
head文件和排序后的临时文件)。
- 使用
总结
这一部分主要完成了以下任务:
- 生成 README 文件,详细说明输出文件的格式和内容。
- 清理临时文件,确保结果文件是有序且整洁的。
第六部分:辅助函数的定义
1. get_hash 函数
sub get_hash {my @info = @_;my %result = ();for my $i (@info) {next if $i =~ /^\s*$/;my ($k, $v) = split(/=/, $i);$result{$k} = $v;}return %result;
}
- 作用:
- 将 Primer3 的输出(以
=分隔的键值对)解析为一个哈希表。 - 遍历输入的每一行,跳过空行。
- 使用
split函数将每一行按=分隔为键和值。 - 将键值对存储到哈希表
%result中。 - 返回解析后的哈希表。
- 将 Primer3 的输出(以
2. get_target_fa 函数
sub get_target_fa {my $id = shift;my $posU = shift;my $posD = shift;if ($posU <= 0) {$posU = 1;}my $seqobj = $ref{$id};my $len = $ref_len{$id};return $seqobj->subseq($posU, $len) if $posD > $len;my $tri = $seqobj->subseq($posU, $posD);return $tri;
}
- 作用:
- 从参考基因组中提取指定区域的序列。
- 参数:
$id:染色体编号。$posU:起始位置。$posD:结束位置。
- 如果起始位置
$posU小于等于 0,则将其设置为 1。 - 使用
Bio::Seq对象$seqobj提取子序列。 - 如果结束位置
$posD超过了染色体长度$len,则提取从$posU到染色体末尾的序列。 - 否则,提取从
$posU到$posD的序列。 - 返回提取的序列。
3. has_ssr 函数
sub has_ssr {my ($id, $seq) = @_;my @SSR;my %typrep = ('2' => '5','5' => '4','3' => '4','6' => '4','1' => '8','4' => '4');my $amb = 200; # interruptions (max_difference_between_2_SSRs)my @typ = sort { $a <=> $b } keys %typrep;my $max_repeats = 1; # count repeatsmy $min_repeats = 1000; # count repeatsmy (%count_motif, %count_class); # countmy ($number_sequences, $size_sequences, %ssr_containing_seqs, %count_motifs); # stores number and size of all sequences examinedmy $ssr_in_compound = 0;my ($nr, %start, @order, %end, %motif, %repeats); # store info of all SSRs from each sequence$seq =~ s/[\d\s>]//g; # remove digits, spaces, line breaks, etc.$id =~ s/^\s*//g; $id =~ s/\s*$//g;#$id =~ s/\s/_/g; # replace whitespace with "_"$number_sequences++;$size_sequences += length $seq;for (my $i = 0; $i < scalar(@typ); $i++) { # check each motif classmy $motiflen = $typ[$i];my $minreps = $typrep{$typ[$i]} - 1;if ($min_repeats > $typrep{$typ[$i]}) { $min_repeats = $typrep{$typ[$i]}; }; # count repeatsmy $search = "(([acgt]{$motiflen})\\2{$minreps,})";while ($seq =~ /$search/ig) { # scan whole sequence for that classmy $motif = uc $2;my $redundant; # reject false type motifs [e.g. (TT)6 or (ACAC)5]for (my $j = $motiflen - 1; $j > 0; $j--) {my $redmotif = "([ACGT]{$j})\\1{" . int($motiflen / $j - 1) . "}";$redundant = 1 if ($motif =~ m/$redmotif/);}next if $redundant;$motif{++$nr} = $motif;my $ssr = uc $1;$repeats{$nr} = length($ssr) / $motiflen;$end{$nr} = pos($seq);$start{$nr} = $end{$nr} - length($ssr) + 1;# count repeats$count_motifs{$motif{$nr}}++; # counts occurrence of individual motifs$motif{$nr}{$repeats{$nr}}++; # counts occurrence of specific SSR in its appearing repeat$count_class{$typ[$i]}++; # counts occurrence in each motif classif ($max_repeats < $repeats{$nr}) { $max_repeats = $repeats{$nr}; };}}if (!$nr) {return (0, \@SSR);} # no SSRs$ssr_containing_seqs{$nr}++;@order = sort { $start{$a} <=> $start{$b} } keys %start; # put SSRs in right ordermy $i = 0;my $count_seq; # countsmy ($start, $end, $ssrseq, $ssrtype, $size);while ($i < $nr) {my $space = $amb + 1;if (!$order[$i + 1]) { # last or only SSR$count_seq++;my $motiflen = length($motif{$order[$i]});$ssrtype = "p" . $motiflen;$ssrseq = "($motif{$order[$i]})$repeats{$order[$i]}";$start = $start{$order[$i]}; $end = $end{$order[$i++]};next;}if (($start{$order[$i + 1]} - $end{$order[$i]}) > $space) {$count_seq++;my $motiflen = length($motif{$order[$i]});$ssrtype = "p" . $motiflen;$ssrseq = "($motif{$order[$i]})$repeats{$order[$i]}";$start = $start{$order[$i]}; $end = $end{$order[$i++]};next;}my ($interssr);if (($start{$order[$i + 1]} - $end{$order[$i]}) < 1) {$count_seq++; $ssr_in_compound++;$ssrtype = 'c*';$ssrseq = "($motif{$order[$i]})$repeats{$order[$i]}($motif{$order[$i + 1]})$repeats{$order[$i + 1]}*";$start = $start{$order[$i]}; $end = $end{$order[$i + 1]};} else {$count_seq++; $ssr_in_compound++;$interssr = lc substr($seq, $end{$order[$i]}, ($start{$order[$i + 1]} - $end{$order[$i]}) - 1);$ssrtype = 'c';$ssrseq = "($motif{$order[$i]})$repeats{$order[$i]}$interssr($motif{$order[$i + 1]})$repeats{$order[$i + 1]}";$start = $start{$order[$i]}; $end = $end{$order[$i + 1]};#$space -= length $interssr}while ($order[++$i + 1] and (($start{$order[$i + 1]} - $end{$order[$i]}) <= $space)) {if (($start{$order[$i + 1]} - $end{$order[$i]}) < 1) {$ssr_in_compound++;$ssrseq .= "($motif{$order[$i + 1]})$repeats{$order[$i + 1]}*";$ssrtype = 'c*';$end = $end{$order[$i + 1]};} else {$ssr_in_compound++;$interssr = lc substr($seq, $end{$order[$i]}, ($start{$order[$i + 1]} - $end{$order[$i]}) - 1);$ssrseq .= "$interssr($motif{$order[$i + 1]})$repeats{$order[$i + 1]}";$end = $end{$order[$i + 1]};#$space -= length $interssr}}$i++;} continue {push @SSR, "$ssrseq";}my $has_ssr = @SSR;return ($has_ssr, \@SSR);
}
- 作用:
- 检测给定序列中是否存在简单序列重复(SSR)。
- 参数:
$id:序列 ID。$seq:待检测的 DNA 序列。
- 使用正则表达式匹配不同类型的 SSR(如单核苷酸重复、二核苷酸重复等)。
- 检测到 SSR 后,记录其位置、重复次数和类型。
- 如果检测到多个 SSR,会检查它们之间的距离是否超过阈值(
$amb),并相应地分类为复合 SSR 或简单 SSR。 - 返回一个布尔值(是否检测到 SSR)和一个包含 SSR 信息的数组引用。
总结
这一部分定义了三个辅助函数:
get_hash:解析 Primer3 的输出,将其转换为哈希表。get_target_fa:从参考基因组中提取指定区域的序列。has_ssr:检测序列中是否存在 SSR,并返回相关信息。
这些函数在脚本的主逻辑中被多次调用,用于处理数据和生成引物设计所需的输入。
相关文章:

indel_snp_ssr_primer
indel标记使用 1.得到vcf文件 2.提取指定区域vcf文件并压缩构建索引 bcftools view -r <CHROM>:<START>-<END> input.vcf -o output.vcf bgzip -c all.filtered.indel.vcf > all.filtered.indel.vcf.gz tabix -p vcf all.filtered.indel.vcf.gz3.准备参…...

图论核心:深度搜索DFS 与广度搜索BFS
一、深度优先搜索(DFS):一条路走到黑的探索哲学 1. 算法核心思想 DFS(Depth-First Search)遵循 “深度优先” 原则,从起始节点出发,尽可能深入地访问每个分支,直到无法继续时回溯&a…...

Java 调用 HTTP 和 HTTPS 的方式详解
文章目录 1. HTTP 和 HTTPS 基础知识1.1 什么是 HTTP/HTTPS?1.2 HTTP 请求与响应结构1.3 常见的 HTTP 方法1.4 常见的 HTTP 状态码 2. Java 原生 HTTP 客户端2.1 使用 URLConnection 和 HttpURLConnection2.1.1 基本 GET 请求2.1.2 基本 POST 请求2.1.3 处理 HTTPS …...

Redis--基础知识点--28--慢查询相关
1 慢查询的原因 1.1 非命令数据相关原因 1.1.1 网络延迟 原因:客户端与 Redis 服务器之间的网络延迟可能导致客户端感知到的响应时间变长。 解决方案:优化网络环境 排查: 1.1.2 CPU 竞争 原因:Redis 是单线程的,…...

目标检测:YOLO 模型详解
目录 一、YOLO(You Only Look Once)模型讲解 YOLOv1 YOLOv2 (YOLO9000) YOLOv3 YOLOv4 YOLOv5 YOLOv6 YOLOv7 YOLOv8 YOLOv9 YOLOv10 YOLOv11 YOLOv12 其他变体:PP-YOLO 二、YOLO 模型的 Backbone:Focus 结构 三、…...

HDFS存储原理与MapReduce计算模型
HDFS存储原理 1. 架构设计 主从架构:包含一个NameNode(主节点)和多个DataNode(从节点)。 NameNode:管理元数据(文件目录结构、文件块映射、块位置信息),不存储实际数据…...

电机控制选 STM32 还是 DSP?技术选型背后的现实博弈
现在搞电机控制,圈里人都门儿清 —— 主流方案早就被 STM32 这些 Cortex-M 单片机给拿捏了。可要是撞上系统里的老甲方,技术认知还停留在诺基亚砸核桃的年代,非揪着 DSP 不放,咱也只能赔笑脸:“您老说的对,…...
.NET 开源工业视觉系统 OpenIVS 快速搭建自动化检测平台
前言 随着工业4.0和智能制造的发展,工业视觉在质检、定位、识别等场景中发挥着越来越重要的作用。然而,开发一个完整的工业视觉系统往往需要集成相机控制、图像采集、图像处理、AI推理、PLC通信等多个模块,这对开发人员提出了较高的技术要求…...

从0到1掌握Kotlin高阶函数:开启Android开发新境界!
简介 在当今的Android开发领域,Kotlin已成为开发者们的首选编程语言。其高阶函数特性更是为代码的编写带来了极大的灵活性和简洁性。本文将深入探讨Kotlin中的高阶函数,从基础概念到实际应用,结合详细的代码示例和mermaid图表,为你呈现一个全面且深入的学习指南。无论你是…...

【OSS】 前端如何直接上传到OSS 上返回https链接,如果做到OSS图片资源加密访问
使用阿里云OSS(对象存储服务)进行前端直接上传并返回HTTPS链接,同时实现图片资源的加密访问,可以通过以下步骤实现: 前端直接上传到OSS并返回HTTPS链接 设置OSS Bucket: 确保你的OSS Bucket已创建…...
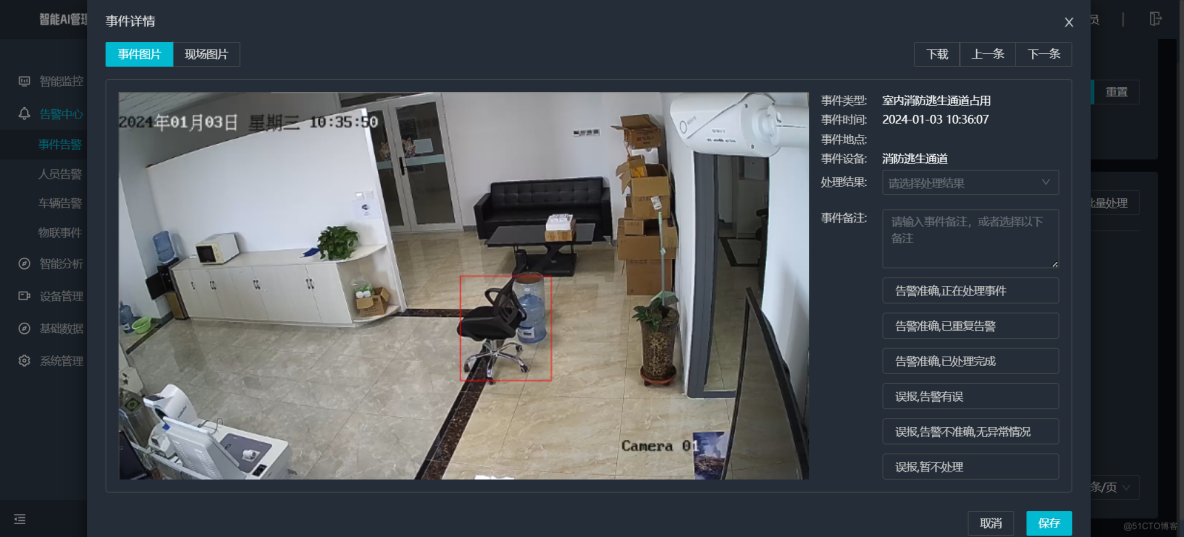
AI智能分析网关V4室内消防逃生通道占用检测算法打造住宅/商业/工业园区等场景应用方案
一、方案背景 火灾严重威胁生命财产安全,消防逃生通道畅通是人员疏散的关键。但现实中通道被占用、堵塞现象频发,传统人工巡查监管效率低、不及时。AI智能分析网关V4结合消防逃生通道占用算法,以强大的图像识别和数据分析能力,…...

商城前端监控体系搭建:基于 Sentry + Lighthouse + ELK 的全链路监控实践
在电商行业,用户体验直接关乎转化率和用户留存。一个页面加载延迟1秒可能导致7%的订单流失,一次未捕获的前端错误可能引发用户信任危机。如何构建一套高效的前端监控体系,实现错误实时追踪、性能深度优化与数据可视化分析?本文将揭…...

Kotlin 中的数据类型有隐式转换吗?为什么?
在 Kotlin 中,基本数据类型没有隐式转换。主要出于安全性和明确性的考虑。 1 Kotlin 的显式类型转换规则 Kotlin 要求开发者显式调用转换函数进行类型转换, 例如: val a: Int 10 val b: Long a.toLong() // 必须显式调用 toLong() // 错…...

基于 HTTP 的邮件认证深入解读 ngx_mail_auth_http_module
一、模块启用与示例配置 mail {server {listen 143; # IMAPprotocol imap;auth_http http://auth.local/auth;# 可选:传递客户端证书给认证服务auth_http_pass_client_cert on;auth_http_timeout 5s;auth_http_header X-Auth-Key "shared_se…...
关于无法下载Qt离线安装包的说明
不知道出于什么原因考虑,Qt官方目前不提供离线的安装包下载,意味着网上各种文章提供的各种下载地址都失效了,会提示Download from your IP address is not allowed,当然目前可以在线安装,但是据说只提供了从5.15开始的…...

Java开发经验——阿里巴巴编码规范实践解析4
摘要 本文主要介绍了阿里巴巴编码规范中关于日志处理的相关实践解析。强调了使用日志框架(如 SLF4J、JCL)而非直接使用日志系统(如 Log4j、Logback)的 API 的重要性,包括解耦日志实现、统一日志调用方式等好处。同时&…...
HTML应用指南:利用GET请求获取全国捞王锅物料理门店位置信息
随着新零售业态的快速发展,门店位置信息的获取变得越来越重要。作为知名中式餐饮品牌之一,捞王锅物料理自2009年创立以来,始终致力于为消费者提供高品质的锅物料理与贴心的服务体验。经过多年的发展,捞王在全国范围内不断拓展门店…...
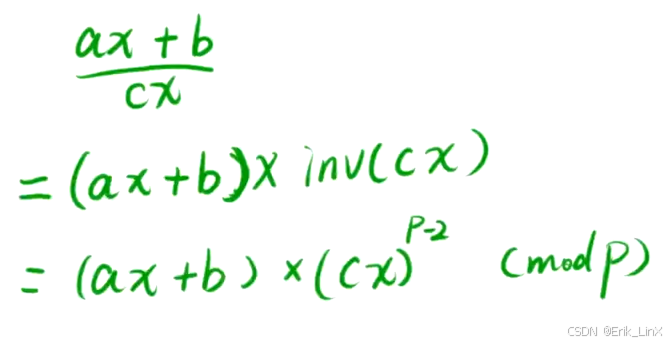
算法日记32:埃式筛、gcd和lcm、快速幂、乘法逆元
一、埃式筛(计算质数) 1.1、概念 1.1.1、在传统的计算质数中,我们采用单点判断,即判断(2~sqrt(n))是否存在不合法元素,若存在则判否,否则判是 1.1.2、假设,此时我们需要求1~1000的所有质数&am…...

黑马点评-分布式锁Lua脚本
文章目录 分布式锁Redis setnxredis锁误删Lua脚本 分布式锁 当我们的项目服务器不只是一台(单体),而是部署在多态服务器上(集群/分布式),同样会出现线程安全问题。不同服务器内部有不同的JVM,每…...

P7-大规模语言模型分布式训练与微调框架调研文档
1. 引言 随着大语言模型(LLMs)在自然语言处理(NLP)、对话系统、文本生成等领域的广泛应用,分布式训练和高效微调技术成为提升模型性能和部署效率的关键。分布式训练框架如 Megatron-LM 和 DeepSpeed 针对超大规模模型…...
机械师安装ubantu双系统:三、GPT分区安装Ubantu
目录 一、查看磁盘格式 二、安装ubantu 参考链接: GPT分区安装Ubuntu_哔哩哔哩_bilibili 一、查看磁盘格式 右击左边灰色区域,点击属性 二、安装ubantu 插入磁盘,重启系统,狂按F7(具体我也忘了)&#…...

ORM++ 封装实战指南:安全高效的 C++ MySQL 数据库操作
ORM 封装实战指南:安全高效的 C MySQL 数据库操作 一、环境准备 1.1 依赖安装 # Ubuntu/Debian sudo apt-get install libmysqlclient-dev # CentOS sudo yum install mysql-devel# 编译时链接库 (-I 指定头文件路径 -L 指定库路径) g main.cpp -stdc17 -I/usr/i…...

kafka学习笔记(三、消费者Consumer使用教程——从指定位置消费)
1.简介 Kafka的poll()方法消费无法精准的掌握其消费的起始位置,auto.offset.reset参数也只能在比较粗粒度的指定消费方式。更细粒度的消费方式kafka提供了seek()方法可以指定位移消费允许消费者从特定位置(如固定偏移量、时间戳或分区首尾)开…...

【后端高阶面经:架构篇】46、分布式架构:如何应对高并发的用户请求
一、架构设计原则:构建可扩展的系统基石 在分布式系统中,高并发场景对架构设计提出了极高要求。 分层解耦与模块化是应对复杂业务的核心策略,通过将系统划分为客户端、CDN/边缘节点、API网关、微服务集群、缓存层和数据库层等多个层次,实现各模块的独立演进与维护。 1.1 …...

网络编程学习笔记——TCP网络编程
文章目录 1、socket()函数2、bind()函数3、listen()4、accept()5、connect()6、send()/write()7、recv()/read()8、套接字的关闭9、TCP循环服务器模型10、TCP多线程服务器11、TCP多进程并发服务器 网络编程常用函数 socket() 创建套接字bind() 绑定本机地址和端口connect() …...
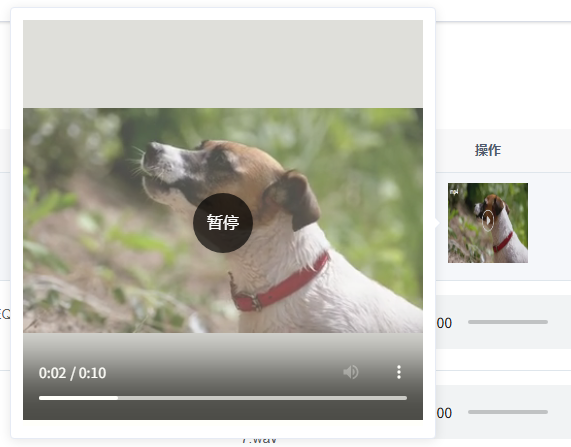
Vue+element-ui,实现表格渲染缩略图,鼠标悬浮缩略图放大,点击缩略图播放视频(一)
Vueelement-ui,实现表格渲染缩略图,鼠标悬浮缩略图放大,点击缩略图播放视频 前言整体代码预览图具体分析基础结构主要标签作用videoel-popover 前言 如标题,需要实现这样的业务 此处文章所实现的,是静态视频资源。 注…...
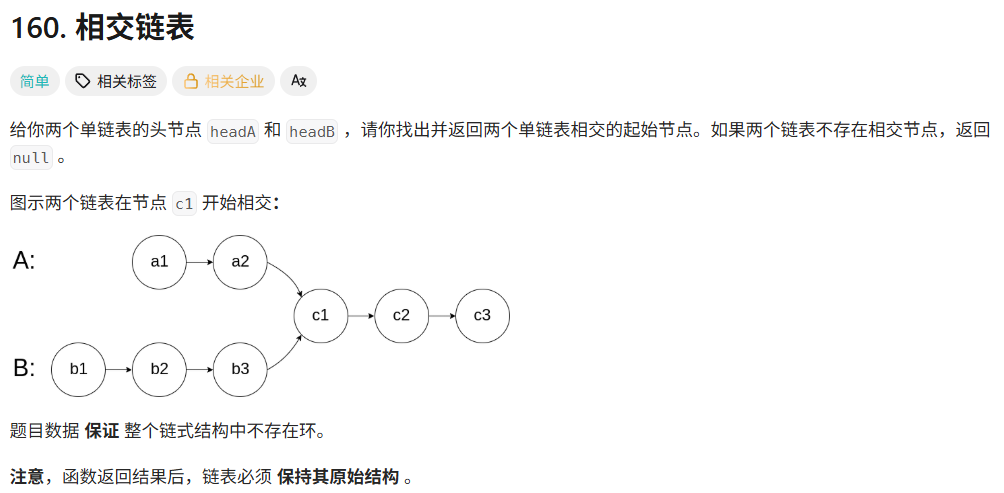
day13 leetcode-hot100-22(链表1)
160. 相交链表 - 力扣(LeetCode) 1.哈希集合HashSet 思路 (1)将A链的所有数据存储到HashSet中。 (2)遍历B链,找到是否在A中存在。 具体代码 /*** Definition for singly-linked list.* pu…...

【Oracle】DQL语言
个人主页:Guiat 归属专栏:Oracle 文章目录 1. DQL概述1.1 什么是DQL?1.2 DQL的核心功能 2. SELECT语句基础2.1 基本语法结构2.2 最简单的查询2.3 DISTINCT去重 3. WHERE条件筛选3.1 基本条件运算符3.2 逻辑运算符组合3.3 高级条件筛选 4. 排序…...
HUAWEI华为MateBook D 14 2021款i5,i7集显非触屏(NBD-WXX9,NbD-WFH9)原装出厂Win10系统
适用型号:NbD-WFH9、NbD-WFE9A、NbD-WDH9B、NbD-WFE9、 链接:https://pan.baidu.com/s/1qTCbaQQa8xqLR-4Ooe3ytg?pwdvr7t 提取码:vr7t 华为原厂WIN系统自带所有驱动、出厂主题壁纸、系统属性联机支持标志、系统属性专属LOGO标志、Office…...
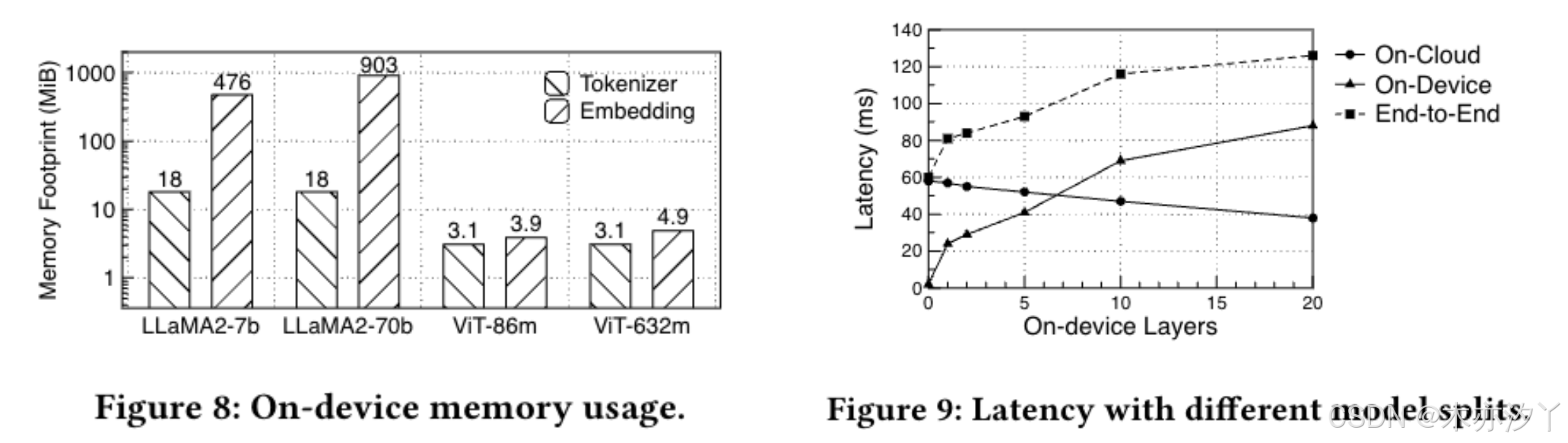
【STIP】安全Transformer推理协议
Secure Transformer Inference Protocol 论文地址:https://arxiv.org/abs/2312.00025 摘要 模型参数和用户数据的安全性对于基于 Transformer 的服务(例如 ChatGPT)至关重要。虽然最近在安全两方协议方面取得的进步成功地解决了服务 Transf…...