论文阅读(九):通过概率图模型建立连锁不平衡模型和进行关联研究:最新进展访问之旅
1.论文链接:Modeling Linkage Disequilibrium and Performing Association Studies through Probabilistic Graphical Models: a Visiting Tour of Recent Advances

摘要:
本章对概率图模型(PGMs)的最新进展进行了深入的回顾,并着重讨论了两个主要的问题:在遗传数据中建立依赖关系模型的基本任务,即连锁不平衡(LD),以及在全基因组关联研究(GWASs)中的下游应用。在整个章节中,所选的例子说明了贝叶斯网络的使用,以及马尔可夫随机域的使用,包括条件和隐马尔可夫随机域。首先,本章介绍了专门用于LD建模的基于PGM方法。下一节将专门讨论基于PGM的GWAS,主要集中在多基因座方法上,其中PGM允许从LD中充分贝内。本节还提供了GWAS中混杂因素确认的说明。下一节将致力于在基因组规模上检测上位性关系。本章以一个总结和一个讨论结束。最后,对今后的工作进行了展望。
关键词:全基因组关联研究,连锁不平衡,上位性关系
本章对概率图模型(PGMs)的最新进展进行了深入的回顾,并着重讨论了两个主要的问题:在遗传数据中建立依赖关系模型的基本任务,即连锁不平衡(LD),以及在全基因组关联研究(GWASs)中的下游应用。在整个章节中,所选的例子说明了贝叶斯网络的使用,以及马尔可夫随机域的使用,包括条件和隐马尔可夫随机域。首先,本章介绍了基于PGM的LD建模方法。下一节将专门讨论基于PGM的GWAS,主要集中在多基因座方法上,其中PGM允许我们从LD中充分贝内。本节还提供了一个说明,以确认在GWAS。下一节将致力于在基因组规模上检测上位性关系。本章以一个总结和一个讨论结束。最后,对今后的工作进行了展望。
9.1介绍
本章建立了一个调查的使用概率图形模型(PGMs)设计用于遗传目的。PGM已广泛用于生物信息学,特别是在DNA序列分析[14],功能基因组学[49]和蛋白质-蛋白质相互作用[31]等领域。在遗传学领域,基因表达分析[13],连锁分析[18]和分子遗传学[23]有助于基于PGM的方法的出现。最近,已经提出了一些成功的方法来破译复杂疾病的遗传因素。本质上,为了满足这一目的,这些方法通常包含遗传数据的建模阶段。在这种背景下,一些研究人员致力于对遗传数据的复杂结构进行建模,这本身就是一个目标。本章重点介绍基于PGM的遗传数据建模和一个主要的下游应用,全基因组关联研究。连锁不平衡(LD)是指一个群体中不同基因座上等位基因的非随机关联。LD主要是由于整个DNA片段从父母传递给后代。变异和突变过程是模糊这种传输方案的两个主要因素。LD检测及其强度、范围和分布的评估是许多遗传学研究的基本步骤。例如,在种群遗传学中,LD模式已被广泛用于研究各种动物和植物种群中涉及的进化和人口统计过程,如混合,迁移和自然选择。
疾病-基因关联是当前基因组学研究的一个热点。关联研究在于确定哪些DNA变异与特定疾病高度相关(即,表型)。特别是单核苷酸多态性(SNP),这是一种流行的遗传标记,广泛用于疾病基因关联研究。检测基因型-表型关联的基本原理如下:影响感兴趣表型的遗传变异在过去的某个时候出现在基因组的一个独特片段上;由于LD,该片段随后与测序变异一起传递给后代;当表型是疾病状态时,预计受检者(病例)和未受检者的基因型(对照)在致病突变周围是不同的,因为病例是祖先携带疾病片段的携带者。由于来自无关个体的染色体经历了比在任何实际大小的系谱中发现的更多的重组事件,关联研究提供了比连锁研究更准确的疾病易感性位点定位,连锁研究处理相关个体(在系谱中)。
高通量基因分型技术的出现带来了识别常见疾病(如高血压、心血管疾病、糖尿病、阿尔茨海默病和各种类型的癌症)的遗传变异的期望[6]。关联研究的两种主要策略已被开发出来:假设驱动[27]和非假设驱动[10]。在本综述章节中,我们只关注非假设驱动的研究。其中,没有特定的区域(基因定位研究),也没有基因或基因组(候选基因研究),被特别怀疑与疾病有关。因此,非假设驱动的研究提出了一个艰巨的计算挑战,因为它们解决了基因组规模,通过定义。全基因组关联研究(GWAS)通常使用蛮力方法来挖掘整个基因组,以寻求推定的关联:例如,基于卡方检验或Cochran-Armitage检验,针对疾病依次测试每个SNP(单SNP方法)[34]。
关联研究可能会受到偏见:人口结构,家庭结构和神秘的相关性是众所周知的混淆因素,导致虚假的协会。在基因组规模上解决混杂因素的存在是必须面对的另一个挑战。另一个挑战是在基因组规模上检测上位性。上位性被定义为两个或多个基因之间的相互作用,以产生感兴趣的表型:对表型的影响不能仅仅通过组合SNP的个体效应来预测。
在一个不确定的框架中,PGM是一个有吸引力的范式来解释高维数据中的依赖关系。一个概率图模型由两部分组成:一个马尔可夫图,表示样本中观察到的变量之间的条件独立性,以及一组概率分布,定义了所谓的参数。每个节点都与一个概率分布相关联:为节点的父变量可以采用的每个特殊值集提供观察节点表示的变量的给定值的概率。在本章中,我们将继续讨论离散变量或离散化变量。由于条件独立性,变量的联合分布分解为更简单的低维分量的乘积。
由于其良好的数学公式和灵活性,PGM通常是多媒体,天文学,文本和Web挖掘以及生物信息学应用等各个领域的首选模型。这个框架允许我们以一种易于处理的方式在复杂的模型上执行计算。必须注意的是,图形模型并不总是明确的。
在这一章中,我们提供了一个全景的最新发展的基础上PGMs和致力于两个主要问题:LD建模的基本任务和GWAS的下游应用。本章的其余部分组织如下:基于PGM的方法致力于LD建模在第9.2节中首次提出;然后下面的部分致力于基于PGM的GWAS,主要集中在多位点方法,其中PGM使我们能够充分贝内于LD。本节还说明了混淆因素的纳入。在第9.4节中,我们回顾了一些基于PGM的方法,致力于在基因组规模的上位性检测。在下一节中,我们概括并讨论所描述的方法的主要优点和缺点。最后,我们指出了未来工作的方向。
9.2连锁不平衡模型
遗传数据的显著之处在于它们显示出高度的相关性。这种相关性定义了一种依赖结构,称为连锁不平衡(LD)。[12,17]的开创性工作描述了人类基因组的基于区块的组织,其中区块代表低单倍型多样性。然后通过Hapmap项目证实了复杂的LD结构[50]。该项目依靠单核苷酸多态性(SNP)对四个参考人群的DNA变异进行了广泛的分析。LD沿基因组沿着变化,显示与重组热点交替的高度相关的SNP区域。后者是短区域,其特征是相关性非常低。简而言之,LD反映了祖先基因组的模糊,主要是但不仅仅是通过重组和突变事件。
然而,这种基于区块的基因组组织并不那么整齐。首先,在一个群体中,每个观察到的单倍型是有限数量的共享短单倍型的镶嵌。尽管如此,镶嵌图中的边界并不是人口中所有科目的共同点:LD是模糊的。其次,已知块之间存在依赖性:已经描述了LD的各种范围[35,41]。通常情况下,我们将反对的模型能够处理长距离LD或集群为基础的模型,以块为基础的模型。
在大多数全基因组关联研究中,数据是通过几个实验室的联合实验获得的。因此,可能发生的是,在收集的整个个体群体中,并非所有基因座都被基因分型。因此,经常需要进行数据插补。为此,选择一个SNP子集,这是suciently信息是第一步,以实现非基因型位点的预测。这种标签SNP的识别与LD的良好模型的可用性有内在联系。除了标签SNP的选择外,进化分析、单倍型推断和基因定位等应用开发LD模型。正如稍后将解释的那样,理想情况下,关联研究(包括全基因组关联研究)也应该利用LD模型。因此,连锁不平衡建模是一个更基本的任务,如上述下游应用的质量要求一个忠实的代表LD。
然而,LD建模是一项复杂的任务。一方面,已知遗传数据表现出强依赖性和弱依赖性,以及紧密SNP之间的依赖性和多位点依赖性,后者的情况对应于LD从短程到长程变化。这样做的结果是LD的模糊性:单倍型块之间的边界不能准确地确定(见图9.1)。另一方面,需要有效的模型学习来代表基因组规模的LD。

概率图模型是一个很有吸引力的框架,可以用来模拟复杂的依赖关系,如LD定义的依赖关系。对于一类特殊的PGM模型,如k阶Markov模型,则不具备所需的灵活性.因此,优选更复杂的PGM。最后,我们将介绍基于PGM的连锁不平衡模型的主要研究方法.马尔可夫随机域(MRF)和贝叶斯网络(BN)代表概率图模型中的两个主要类别.这两个类别都被研究过,以模拟连锁不平衡。我们首先提供了一个总体概况,给出了建议的主要特点。然后,对模型进行了详细的描述。本文讨论了这些模型的优点和注意事项,重点讨论了各种标准,包括潜在变量的存在、基于块的建模与基于集群的建模以及可扩展性。最后一个小节概括了这些方法。
9.2.1总体全景
托马斯和坎普是第一个估计离散概率图模型来计算等位基因频率联合分布的人[54]。第一种方法依赖于可分解的马尔可夫随机场(DMRF)的一般类别。由于没有可分解属性的操作表征,强制性的尝试和测试策略严重阻碍了可扩展性。为了避免这个问题,托马斯的进一步工作依赖于导航MRF的限制类,称为DMRF:这个限制类基于区间图(IG)[52,53]。设计保证现任IG图G的邻居G也是IG的操作是简单的。然而,这种效率是有代价的-DMRF对基于IG的DMRF的限制。因此,最近Abel和托马斯再次关注DMRF类[1]。这一次,由于来自图论的属性,获得了可扩展性。在可分解的马尔可夫随机场中,变量的因子化联合分布的分量是团,而这些分量是贝叶斯网络中的模型顶点。因此,贝叶斯网络似乎比可分解的马尔可夫随机分布更自然、更灵活和更可解释的工具。最后,贝叶斯网络的联合分布通过有向无环图(DAG)进行编码,这适合于与因果关系相关的任何进一步解释的尝试。
Villanueva和Maciel选择通过贝叶斯网络对LD进行建模,因为他们有兴趣了解基因座之间关联的因果关系[60]。与之前的方法一样,Lee和Shatkay也采用贝叶斯网络对LD进行建模[30]。除了一般的贝叶斯网络之外,所有其他基于BN的解决LD建模的建议都共享潜变量(LV)的共同使用。Zhang和Ji描述了一个双层贝叶斯网络,它是(独立的)潜在类模型的集合[61]。也是一个两层模型,Nean的嵌入式贝叶斯网络比前一个模型更复杂[40]。上述四种基于BN的方法的共同缺点是缺乏可扩展性。相反,Mourad等人的潜在树模型森林(FLTM),另一个基于BN的具有潜在变量的建议是满足可扩展性标准的多层分层模型[37]。
9.2.2可分解马尔可夫随机场
托马斯与多位合著者一起,通过马尔可夫随机场对LD建模领域做出了很大贡献。这导致了不少于四种学习算法的建议,其中两种专注于可以用区间图(IG)表示的DMRF子类。
前部工程
可分解图允许在给定数据的情况下有效地计算结构的可能性。因此,可以在优化基于对数似然的分数的同时导航结构空间来执行结构学习。在[54]中,为了探索DMRF空间,设计了基于随机选择的节点的连接或断开的操作来构建现任图G的邻域。无论用于搜索空间导航的方法(下坡搜索或模拟退火),一个严重的问题降低了模型学习算法的效率:一般可分解图的操作特征不可用。因此,从现任图G开始,必须检查后验概率是否建议G在G的邻域中是可分解的。因此,托马斯和坎普的第一个模型是不可扩展的,因为在学习算法中,大量的时间被浪费在检查图的可分解性和拒绝不相关的结构上。
为了提高效率,托马斯接下来考虑了DMRF的一个子类,即可以用区间图(IG)表示的DMRF。提高模型学习效率的关键是替换IG表示采样到图采样。一个图可以用区间图表示,当且仅当它的顶点可以对应于真实的直线上的区间,使得两个顶点连接,只要它们对应的区间重叠。在最早的基于IG的提议[52]中,IG搜索空间通过简单的操作进行导航。一个例子是随机选择的间隔长度的变化。为了进一步降低时间复杂度,第二个基于IG的版本要求任何间隔在每一侧的扩展不超过最大值[53]。
返回到一般可分解马尔可夫随机场

9.2.3无潜在变量的贝叶斯网络方法
基于分数与约束的数据依赖性确认

模型后处理

9.2.4基于贝叶斯网络的潜在变量方法
基于两层BN的方法
Zhang和Ji [61]的双层贝叶斯网络属于利用关于单倍型块结构的知识的模型的类别,如下所示:在群体中观察到的单倍型倾向于局部聚类成相似单倍型的组;因此,聚类的数量以及因此它们的大小沿基因组沿着变化;建模的原理在于将每个位点分配给一个簇,每个簇被表示为一个潜在类模型(见图9.2)。潜在类模型(LCM)是一个BN,其中一个唯一的潜在变量连接到它的每个子变量,子变量彼此独立,以它们的潜在父变量为条件。在这一类中,Zhang和Ji的方法改进了Kimmel和Shamir的方法,其中模型也是LCM的集合,但基于块。在基于块的模型中,簇必然包含基因组上的物理延伸,而在最灵活的基于簇的模型中,来自遥远区域的SNP可以被允许在同一簇中。为了在缺失或潜在变量的情况下学习贝叶斯网络,标准策略包括运行结构期望最大化(SEM)过程。SEM在于连续地优化以结构为条件的参数,然后优化以参数为条件的结构。

在Zhang和Ji的简单模型中,长程LD和近SNP之间的LD被同等地表示。与此相反,Nean [40]的嵌入式贝叶斯网络允许将长程LD表示为高阶相关性(见图9.3)。同样是一个两层模型,宁安的模型比张和季的模型更复杂,因为构成第一层的观察变量之间可能存在依赖关系(但前提是后者共享同一个父变量--潜变量)。同时,允许上层中的潜在变量之间的依赖性。适用于这种嵌入式结构的构造的算法自然是SEM递归过程:首先执行标准SEM过程的独立运行以学习作为扩充LCM的局部结构(即,子节点可以在LCM内连接);一旦被输入,扎根于局部结构的潜在变量就扮演全局结构的观察变量的角色;然后,最终的SEM步骤学习该全局结构。在张、季的方法和聂安的方法中,结构方程的结构学习步骤实现了爬山。Zhang和Ji所实现的移动是LCM之间的成员资格改变(即,SNP从一个LCM移动到另一个LCM)。Nean使用标准的移动(增加,删除,或反转一个边缘).Nean模型的复杂性是以减轻计算负担为代价的,对局部结构施加了强约束:基因组被分成固定共同大小的连续窗口,并从这些块中构建尽可能多的LCM。[61]和[40]中的模型之间的根本区别在于,第一个模型是基于簇的,而第二个模型是基于块的。后一种方法缺乏灵活性是一个严重的缺点。Zhang和Ji的基于群集的方法处理不同大小的群集。然而,尽管有了这种改进,缺点在于必须指定簇的数量。

可扩展的分层方法
Mourad等人的分层模型,FLTM(潜在树森林模型)[37]是张和季的模型的推广,其中只允许一层潜在变量(见图9.4)。潜在树模型(LTM)可以被描述为嵌入式LCM的集合,从而定义潜在变量的层。由于成对依赖和多位点LD可以同时表示在层次结构中,FLTM允许比其他方法更忠实的遗传数据建模。特别是,层次结构提供了一个灵活的模型,能够最好地反映LD的模糊性。此外,这种层次结构是满足基因组规模可扩展性要求的关键。

用于学习FLTM的自适应上升分层聚类过程如下处理数据:在每个凝聚步骤中,使用分区方法来识别成对依赖变量的集团;每个这样的集团旨在通过LCM被归入潜在变量。对于每个LCM,执行参数学习(通过标准的期望最大化(EM)算法),这会产生潜在变量的边际分布和子变量的条件分布。然后,进行(线性)概率推断,以填补相应的潜在变量的缺失数据。在LTM森林中,只有那些对所包含的变量有足够信息的潜变量必须被保留。概括地说,学习过程的三个主要组成部分是集团划分,基于LCM的构建和基于LCM的估算。
经验复杂性
上述两层BN模型都不是可扩展的。例如,Nean [40]不仅使用通过连续窗口实现的分治策略,她还采用并行化。基于窗口的方法也被用于Mourad和同事的方法的第一个版本[37]。然而,与Nean的提议相反,这个第一个版本在没有诉诸并行化的情况下达到了可扩展性。后来,这种移动窗口过程被更令人满意的滑动窗口方法所取代:与托马斯及其同事的工作[1,53]相同,基因组上相距太远的SNP不允许在同一集团中。第一个版本可以在大约15小时内处理描述2000个个体的10个SNP的数据集,任意窗口大小为100个SNP [36]。当将滑动窗口大小设置为0.5 Mbp(捕获LD的合理选择)时,他们的新算法运行时间不到12小时[46,47]。必须强调的是,由于这个新版本在10次重启后运行EM,因此相对于初始版本有了显着的改进。最后,复杂度大致为O(p),其中p表示SNP的数量;复杂度也是滑动窗口大小的线性函数。应用软件CFHLC+(层次潜在类模型森林的构建)可在http://sites.google.com/site/raphaelmourad/Home/programmes上获得。
FLTM模型是张和纪模型的简单性与Nean模型的复杂性之间的折衷,由于其层次结构,FLTM模型是唯一可扩展的方法。如[39]中所强调的,施加分层结构是减轻计算复杂度的关键。与以前描述的所有基于PGM的方法来建模LD相比,局部搜索不再是规则;相反,实现了一个特定的过程,贪婪的,迭代的,自底向上的变量聚类。对BN结构的这种约束不是对LD建模精度的警告,因为LD本质上是分层的。
9.2.5重述
表9.1概括了描述LD的各种模型及其学习算法的主要特征。LD建模的动机表明每种方法:单倍型推断,关联块的识别,和选择标记SNP。在这些提议中,只有三个是可扩展的。在两种情况下,通过使用特定的数据结构,托马斯等人的基于区间图的马尔可夫随机场和Mourad等人的分层贝叶斯网络,可以获得可扩展性。因此,可以设计特定的学习算法来构建这些约束结构。然而,基于块的建模方法,如基于区间图的方法,仍然是LD的粗略近似。相比之下,Mourad等人的分层FLTM模型是一种基于聚类的方法。Abel和托马斯最近的工作也提供了一个可扩展的基于集群的方法,这要归功于对图论属性的利用。前两种方法之间的主要区别在于Abel和托马斯的核心模型学习过程处理分阶段数据。因此,这个核心过程被合并到一个更大的框架中,交替模型学习和阶段推理。因此,Abel和托马斯的方法也解决了单倍型推断。单倍型推断不是基于FLTM策略的目标。最后,这两种方法的一个共同特点是采用最大物理距离来控制运行时间。

9.3全基因组关联研究的单SNP方法
本节回顾了概率图形模型,旨在实现全基因组关联研究(GWAS)的情况下,可能有几个独立的未知致病遗传因素被怀疑在疾病的易感性:每个致病SNP在每个基因的影响疾病施加一个小的加性效应的疾病表型。这种情况与相互作用因素的情况相反,我们认为这是本章上下文中对上位性的定义。在最简单的设计中,单独测试任何SNP对疾病的影响,从而指定单SNP方法。最基本的GWAS将依次考虑每个SNP,并针对所研究的表型进行测试。然而,当怀疑混杂因素干扰基因型以影响表型时,模型必须整合这些协变量。更复杂的方法将充分利用LD的知识,表现出过量的单倍型共享的情况下,周围的致病基因座,这些被称为多位点的方法。
在我们集中于全基因组范围之前,我们布里伊报告了一些致力于基因定位和候选基因研究的工作。在[43]中,使用随机重新开始的贪婪搜索来学习存在协变量(如种族和胆固醇水平)的贝叶斯网络。在[45]中,所采用的K2程序使用依赖于以下变量排序的启发式:较旧的SNP作为较新SNP的子代进行测试。前SNPs的特征在于其变体在人群中的分布比最近的SNPs更均匀。最后,另一个重要的工作是将上述托马斯的方法扩展到发现SNP-表型关联[51]。
然而,毫不奇怪,所有最近涉及概率图形模型的工作都建立在基因组规模的易处理性基础上。此外,概率图形模型是一个理想的框架,以整合各种来源的数据,以加强可信度的结果。在本节中,我们首先描述了一种用于解决混杂因素的方法。然后,我们描述了三个开发用于实现多位点GWAS的模型。这一节将特别展示两种关于马尔可夫随机域的变化:隐马尔可夫随机域和条件随机域.此外,还将描述与图形模型紧密相连的模型--变长马尔可夫链。
9.3.1混杂因素的整合



9.3.2 GWAS多位点方法
在第9.3节的介绍中,我们提到了单SNP方法的替代方法:在多位点方法中,目的是通过有关连锁不平衡的知识,加强对推定的单位点与疾病之间关联的评估。也就是说,关联知识预计将从LD中的多个SNP与给定的SNP得到加强。连锁不平衡很可能揭示了一个致病基因座周围的单倍型共享过量,病例。在本小节中,我们将首先回到布里-伊模型的托马斯[54],一方面,穆拉德等人。我们将证明,为了GWAS的目的,对这些模型进行后处理是很简单的。然后,我们将介绍Verzilli等人的方法。[59],该方法基于可分解的随机马尔可夫链。将描述马尔可夫链模型的一个变体,该变体是布朗宁和布朗宁的金标准方法的基础[9]。另一方面,隐马尔可夫随机场也被研究用于GWAS目的:我们将描述Li等人的方法。
连锁不平衡的后处理模型
在小节9.2.2(第9.2.2页)中,我们介绍了Thomas及其合作者的工作,他们率先使用可分解马尔可夫随机场(DMRFs)来模拟连锁不平衡[54, 52, 53, 1]。
当用于捕捉连锁不平衡时,DMRF模型还允许测试疾病关联。实现这一目标的一个简单方法是在DMRF中包含一个代表表型(疾病状态指标)的顶点。然后,可以将估计的模型与通过移除表型和位点顶点之间任何链接获得的子模型进行比较。为此目的,可以使用标准的卡方似然比检验。
Mourad等人指出,他们的FLTM层次模型对于GWAS目的是相关的:从位于层次结构高层的节点(即,潜在变量)开始,一种最佳优先搜索策略将通过基因组中越来越窄的区域,以确定可能与疾病相关的区域。一个严重的缺陷可能是潜在变量对其子变量的信息不足,特别是在层次结构的高层,这将导致能力不足。为了证明层次结构中的自下而上的信息衰减可以被控制以提供可靠的模型,Mourad等人进行了密集测试[47]:在36种条件下模拟了单SNP-表型关联。所有潜在变量都接受了与表型的关联测试。所有是因果SNP祖先的节点都成功地捕获了与表型的间接关联。相比之下,其他潜在节点总体上显示出非常弱的关联。因此,识别这样的祖先节点将允许指出潜在的因果标记,作为以这个祖先节点为根的树的叶节点。
通过模型平均评估关联

通过变长马尔可夫链的GWAS

![]()
![]()

![]()
![]()



在该方法中,多重测试问题通过排列测试来解决。在整个排列中保持相同的单倍型局部簇集。该方法的实现可作为由包Beagle(http://faculty.washington.edu/browning/beagle/beagle.html)提供的多个软件程序之一获得。由于快速建模,该方法被称为GWAS中为数不多的可扩展多位点方法之一。
通过隐马尔可夫随机场的GWAS
多位点方法利用了这样一个事实,即如果几个SNP都与真实的疾病变体连锁不平衡,则这种依赖性预计会增加检测与疾病相关的SNP的能力。在[32]中,Li等人提出将LD信息正式纳入贝叶斯过程,该过程旨在估计给定SNP与感兴趣疾病相关的后验概率。关键是,这种后验概率不仅取决于在该SNP处观察到的基因型数据,而且还取决于与该特定SNP处于强LD的基因型。Li等人使用的框架是一个隐马尔可夫随机场。


9.3.3优势和局限性
在本小节中,我们首先强调了这两种方法之间的差异,主要集中在用于确定假定关联的策略和用于评估关联的重要性的程序上。最后,我们通过插图表明在何种程度上的方法是可扩展的。
寻找关联
已经提出了四种方法,解决多位点GWAS分析。所有四种方法都利用连锁不平衡模型。在其类别中,Huang及其合作者的方法是为了科普混杂因素而开发的,并专注于个体表型之间的相似性。在托马斯等人和Verzilli及其同事的方法中,马尔可夫随机场模型中的集团是SNP高度相关的区域。因此,SNP和代表表型的节点之间的任何边缘都可以确定疾病与该SNP所定义的区域的推定关联,以及与该SNP具有强LD的SNP。的方法,后处理的模型是快速的(似然比的模型与和没有边缘具有作为端点的表型)。在Verzilli et al.在该方法中,进行模型平均:关联的后验概率被估计为连接给定SNP与表型顶点的边的存在的百分比,并且计算贝叶斯因子。在所描述的其他图形模型中,表型不作为节点被包括。在Mourad et al.的潜在树森林模型(FLTM),潜在变量支持相同的解释,在上述两种方法的集团,即区域的SNPs处于强连锁不平衡。布朗宁和布朗宁[7]的可变长度马尔可夫链方法采用局部单倍型聚类;这样的单倍型聚类可以被视为潜在变量。在RMF、FLTM和VLMC中,单倍型的团、树和局部簇的组织分别解释了连锁不平衡的原因。
此外,Mourad等人以及布朗宁和布朗宁的模型被开发来降低GWAS(或任何其他下游分析)中的数据维数:这些模型允许我们分别对(有限数量的)潜在变量和局部单倍型进行关联检验。尽管如此,FLTM和VLMC模型之间的一个主要区别仍然是前者是基于簇的,而后者是基于块的。最后,FLTM模型由于其层次结构而表现出最高程度的灵活性.这种基于树的结构是唯一一种可以同时模拟近距离和远距离依赖性的结构。在Li等人s的方法,LD模式随机地确定了与疾病相关的(潜在)指示符(His)之间的依赖性模式。无论是Li等人'的方法和Verzilli等人的方法。的方法使用抽样过程来处理关联的发现。Verzilli等人'的GWAS方法依赖于模型平均。Li等人操作的模型后处理对指示剂His的分布进行采样,以估计每个SNP的无关联的后验概率。
SNP的显著性评估

经验复杂性
在利用连锁不平衡模型的所有上述方法中,通过结合一些约束来减轻计算负担:布朗宁和布朗宁设定条件以控制可变长度马尔可夫链的大小(宽度)。在所述的其他三种方法中,在相同的集团中允许成对依赖的SNP,但要服从最大物理距离(托马斯等人,Verzilli等人,Mourad等人)。在Mourad等人的递归模型中,潜在变量也受到该约束。此约束涉及LD范围。在VLMC方法中,对VLMC宽度设置的条件也间接地解释了LD范围,尽管理解不如其他方法直观。
除了评估他们的方法的功效之外,例如在描述3400个个体的360 657个SNP的真实的数据集上,Huang等人分析了他们的方法可扩展的程度。为了进行比较,线性混合模型在33小时内运行了7579个SNP和37830个个体的基准。相比之下,对于相同的数据集,基于CRF的算法的执行时间为48分钟。重要的是,由于未来的研究往往涉及数万人,预计会有更大的加速。Verzilli及其同事的方法的可扩展性已经在描述1018个个体的100 000个SNP的基准上进行了检查。在这种情况下,只需要4分钟。Mourad等人'的最新版本显示,运行12小时的10万标记和2000人。已知布朗宁和布朗宁的比格犬金标准处理大规模遗传数据集,该数据集具有对数千个个体进行基因分型的数十万个标记(没有报告运行时间)。比格犬的标记数量呈线性的原因是,当决定是否在某个给定水平上合并两个节点时,不必一直观察到染色体的末端。最后,Li和同事的方法是唯一一种通过使用连续窗口来扩展到全基因组数据的方法。例如,描述3075例病例和对照的30216个SNP被分成1000个SNP的组(没有报告运行时间)。
概括
表9.2概括了本节中描述的基于PGM的方法,这些方法专门用于单个SNP-表型关联。

9.4在基因组尺度上识别上位性
在本上下文中,上位性一词用于表示不同遗传基因座对所研究表型的影响偏离独立性:因此,不能通过组合单个基因座的影响来直接预测表型的影响[11]。参与这种遗传相互作用的单个基因座通常产生微弱的边缘影响,如果不是甚至可以忽略不计的话。概率图模型自然地提供了一个有吸引力的框架来检测变量之间的相互作用。在报告了Jiang及其合作者[26]提出的基于BN的一般方法之后,本节介绍了Han和Chen [20]以及Jiang等人[25,26]围绕同一艾德贝叶斯模型的变化。然后,描述了Han等人[21]的基于Markov毯的方法。
9.4.1贝叶斯网络方法




9.4.2基于马尔可夫毯的方法



9.4.3重述
表9.3概括了上述三种方法的特征。

一方面,Han和Chen的方法,另一方面,Jiang等人的MBS方法,依赖于相同的简化的艾德贝叶斯网络模型。为了避免复杂性问题,Han和Chen根据它们的边际效应预先选择SNP。然而,可以证明存在一些三方互动。相比之下,为了降低复杂性,Jiang等人将多途径相互作用限制为至少包含双向上位相互作用。与Han和Chen在基因组规模上实施的两阶段策略相比,在Jiang等人的方法中,上位性模式不需要由对疾病有边缘效应的SNP组成。然而,很难估计哪种上位模式更常见:包含双向上位相互作用的上位模式或所有SNP对疾病具有个体效应的上位模式。
最后,必须回顾一下,Jiang等人的MBS方法并没有学习整个贝叶斯网络,而是学习一个简化的艾德DDAG网络。更进一步,Han等人的基于Markov Blanket的方法避免了学习整个贝叶斯网络的结构。因此,尽管在基因组规模上必然是复杂的,但这种方法似乎很有吸引力,可以在这种规模上检测高阶上位模式。此外,参数学习是短路的。在[20]和[21]中可以分析相同的数据集。然而,基于马尔可夫毯的方法不需要预选排名靠前的SNP。
9.5讨论
通过概率图模型对连锁不平衡进行建模的方法是经验性的。大多数情况下,他们不依赖于群体遗传学的概念:基于PGM的方法经验性地估计由重组过程、突变过程和群体历史引起的基因座之间的独立性和依赖性,而不对任何这些进行建模。对应的是,基于PGM的模型不能直接用于推断种群如何进化或未来将如何进化。

在机器学习领域,贝叶斯网络是在不确定框架中表示(直接和间接)依赖关系以及条件独立性的领先架构。铂族金属使我们能够建立良好的近似的潜在现实。因此,它们是下游分析的有力工具。最后,虽然从高维数据中学习模型通常是一个问题,但几个基于PGM的作品已经成功地设计了用于分析基因组规模数据的可扩展算法。
9.6观点
最近,在基因组尺度上使用概率图形模型取得了进展。然而,仍有取得进展的余地。
随着基因分型技术成本的不断降低,最终目的是将该方法应用于具有数十万或数百万个基因座和大量基因分型个体(数万个)的大量基因组区域。因此,研究人员将不得不检查到目前为止获得的可扩展性是否仍然适用于通过荟萃分析输出或收集的更多海量数据。很可能需要设计高度可扩展的算法。在这条线上,结构学习程序必须量身定制。它已经表明,特定的程序,如那些基于图论或变量的迭代层次聚类允许我们实现基因组规模的方法。利用这一事实,即连锁不平衡主要涉及密切的SNPs开辟了道路,稀疏的图形模型的调查。在这方面,基于LASSO的方法等技术很有前途[15]。
毫无疑问,最具挑战性的问题之一,由于其高度的组合方面,上位性的检测仍处于起步阶段。方法基于Markov Blanket概念对整个贝叶斯网络进行短路学习。然而,复杂性仍然很高。在这一领域有迫切的需要,因为研究人员现在认为上位性可能是疾病遗传风险的暗物质的重要部分。
在组学数据整合的时代,生物学先验信息的整合有望提高模型的准确性和学习这些模型的算法的效率。例如,当明确考虑SNP之间的成对相关性时,如在Li和合作者的模型中,将值得将关于蛋白质-蛋白质相互作用的额外信息整合到分析中。一个多尺度的方法将首先考虑一个给定基因内SNP的加权LD图。然后,关于基因网络或蛋白质-蛋白质相互作用网络的知识将提供一个大的组合SNP网络。更一般地说,整合与研究中的表型相关的合理基因调控网络的先验信息,肯定会有助于限制可能的依赖性空间。无论使用哪种模式,数据集成都是一种有前途的解决方案。概率图形模型被认为非常适合于整合异质性数据的目的:因此,遗传学、基因表达和蛋白质组学可以整合在一个艾德框架中。由于这一灵活性,PGM有望为改善疾病的预防、诊断与治疗铺平道路。
参考文献
略
相关文章:
论文阅读(九):通过概率图模型建立连锁不平衡模型和进行关联研究:最新进展访问之旅
1.论文链接:Modeling Linkage Disequilibrium and Performing Association Studies through Probabilistic Graphical Models: a Visiting Tour of Recent Advances 摘要: 本章对概率图模型(PGMs)的最新进展进行了深入的回顾&…...

python小知识-typing注解你的程序
python小知识-typing注解你的程序 1. Typing的简介 typing 是 Python 的一个标准库,它提供了类型注解的支持,但并不会强制类型检查。类型注解在 Python 3.5 中引入,并在后续版本中得到了增强和扩展。typing 库允许开发者为变量、函数参数和…...

git基础使用--1--版本控制的基本概念
git基础使用–1–版本控制的基本概念 1.版本控制的需求背景,即为啥需要版本控制 先说啥叫版本,这个就不多说了吧,我们写代码的时候肯定不可能一蹴而就,肯定是今天写一点,明天写一点,对于项目来讲ÿ…...

“新月智能武器系统”CIWS,开启智能武器的新纪元
新月人物传记:人物传记之新月篇-CSDN博客 相关文章链接:星际战争模拟系统:新月的编程之道-CSDN博客 新月智能护甲系统CMIA--未来战场的守护者-CSDN博客 “新月之智”智能战术头盔系统(CITHS)-CSDN博客 目录 智能武…...
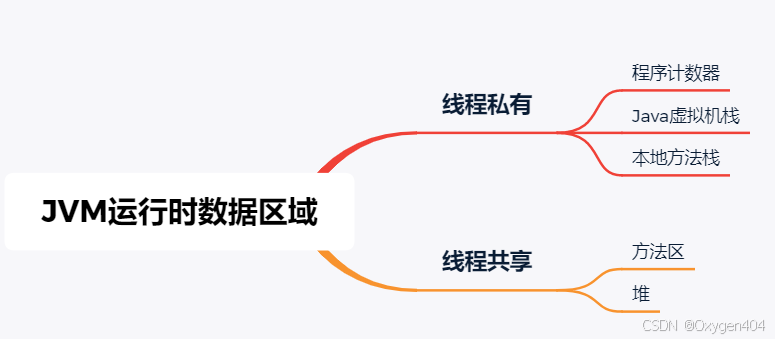
JVM运行时数据区域-附面试题
Java虚拟机在执行Java程序的过程中会把它所管理的内存划分为若干个不同的数据区域。这些区域 有各自的用途,以及创建和销毁的时间,有的区域随着虚拟机进程的启动而一直存在,有些区域则是 依赖用户线程的启动和结束而建立和销毁。 1. 程序计…...
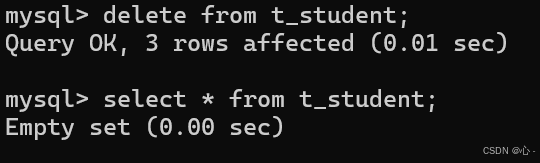
增删改查(CRUD)操作
文章目录 MySQL系列:1.CRUD简介2.Create(创建)2.1单行数据全列插入2.2 单行数据指定插入2.3 多⾏数据指定列插⼊ 3.Retrieve(读取)3.1 Select查询3.1.1 全列查询3.1.2 指定列查询3.1.3 查询字段为表达式(都是临时表不会对原有表数据产生影响)…...

Vue.js `Suspense` 和异步组件加载
Vue.js Suspense 和异步组件加载 今天我们来聊聊 Vue 3 中的一个强大特性:<Suspense> 组件,以及它如何帮助我们更优雅地处理异步组件加载。如果你曾在 Vue 项目中处理过异步组件加载,那么这篇文章将为你介绍一种更简洁高效的方式。 什…...
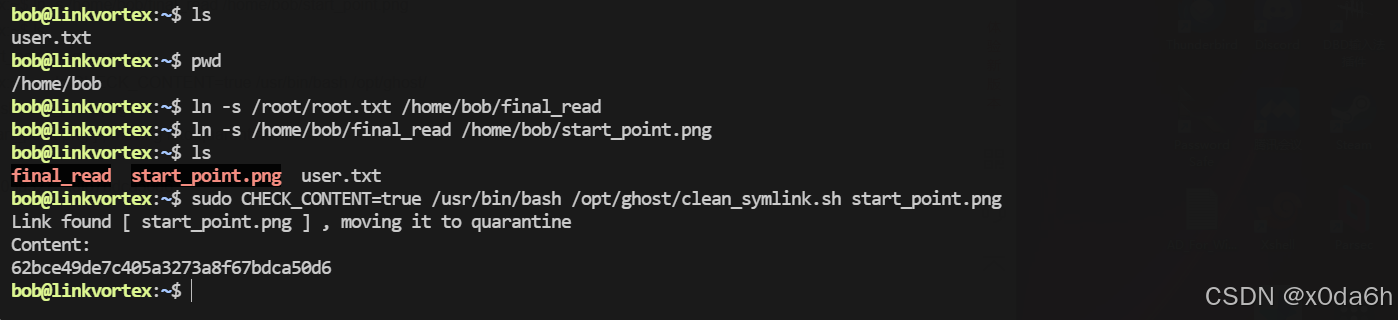
HTB:LinkVortex[WriteUP]
目录 连接至HTB服务器并启动靶机 信息收集 使用rustscan对靶机TCP端口进行开放扫描 使用nmap对靶机TCP开放端口进行脚本、服务扫描 使用nmap对靶机TCP开放端口进行漏洞、系统扫描 使用nmap对靶机常用UDP端口进行开放扫描 使用gobuster对靶机进行路径FUZZ 使用ffuf堆靶机…...
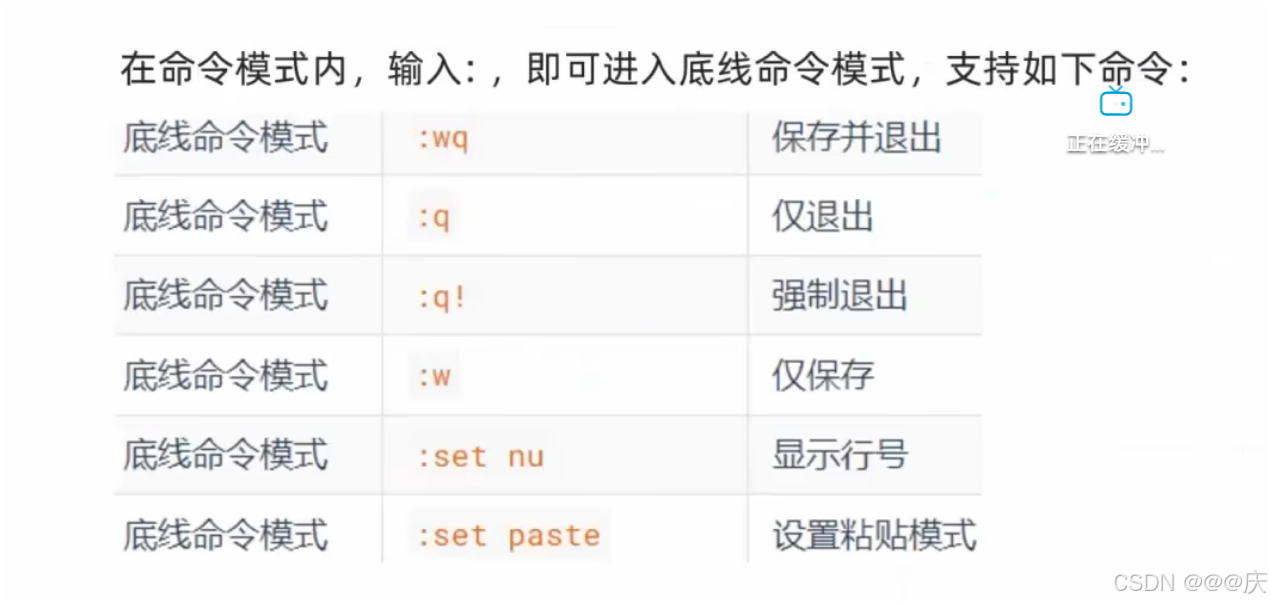
Linux命令入门
Linux命令入门 ls命令 ls命令的作用是列出目录下的内容,语法细节如下: 1s[-a -l -h] [Linux路径] -a -l -h是可选的选项 Linux路径是此命令可选的参数 当不使用选项和参数,直接使用ls命令本体,表示:以平铺形式,列出当前工作目录下的内容 ls命令的选项 -a -a选项&a…...
【问题】Chrome安装不受支持的扩展 解决方案
此扩展程序已停用,因为它已不再受支持 Chromium 建议您移除它。详细了解受支持的扩展程序 此扩展程序已停用,因为它已不再受支持 详情移除 解决 1. 解压扩展 2.打开manifest.json 3.修改版本 将 manifest_version 改为3及以上 {"manifest_ver…...
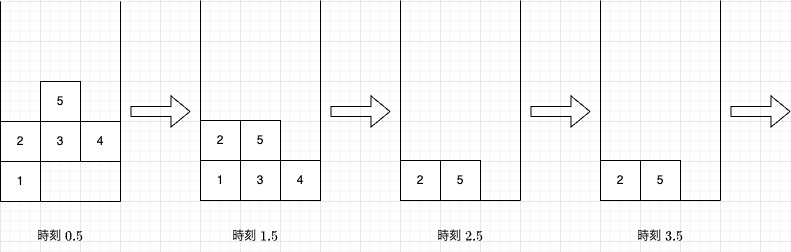
【题解】AtCoder Beginner Contest ABC391 D Gravity
题目大意 原题面链接 在一个 1 0 9 W 10^9\times W 109W 的平面里有 N N N 个方块。我们用 ( x , y ) (x,y) (x,y) 表示第 x x x 列从下往上数的 y y y 个位置。第 i i i 个方块的位置是 ( x i , y i ) (x_i,y_i) (xi,yi)。现在执行无数次操作,每一次…...
使用 SpringBoot+Thymeleaf 模板引擎进行 Web 开发
目录 一、什么是 Thymeleaf 模板引擎 二、Thymeleaf 模板引擎的 Maven 坐标 三、配置 Thymeleaf 四、访问页面 五、访问静态资源 六、Thymeleaf 使用示例 七、Thymeleaf 常用属性 前言 在现代 Web 开发中,模板引擎被广泛用于将动态内容渲染到静态页面中。Thy…...

【Java异步编程】CompletableFuture综合实战:泡茶喝水与复杂的异步调用
文章目录 一. 两个异步任务的合并:泡茶喝水二. 复杂的异步调用:结果依赖,以及异步执行调用等 一. 两个异步任务的合并:泡茶喝水 下面的代码中我们实现泡茶喝水。这里分3个任务:任务1负责洗水壶、烧开水,任…...

Nginx知识
nginx 精简的配置文件 worker_processes 1; # 可以理解为一个内核一个worker # 开多了可能性能不好events {worker_connections 1024; } # 一个 worker 可以创建的连接数 # 1024 代表默认一般不用改http {include mime.types;# 代表引入的配置文件# mime.types 在 ngi…...

Unity开发游戏使用XLua的基础
Unity使用Xlua的常用编码方式,做一下记录 1、C#调用lua 1、Lua解析器 private LuaEnv env new LuaEnv();//保持它的唯一性void Start(){env.DoString("print(你好lua)");//env.DoString("require(Main)"); 默认在resources文件夹下面//帮助…...

AI-ISP论文Learning to See in the Dark解读
论文地址:Learning to See in the Dark 图1. 利用卷积网络进行极微光成像。黑暗的室内环境。相机处的照度小于0.1勒克斯。索尼α7S II传感器曝光时间为1/30秒。(a) 相机在ISO 8000下拍摄的图像。(b) 相机在ISO 409600下拍摄的图像。该图像存在噪点和色彩偏差。©…...
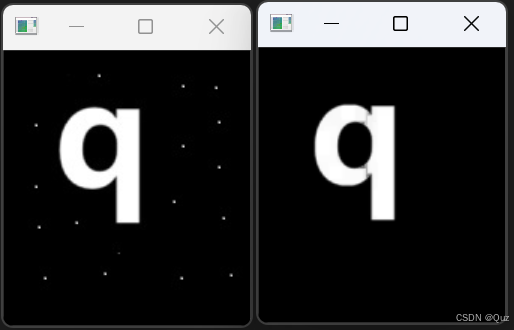
OpenCV:开运算
目录 1. 简述 2. 用腐蚀和膨胀实现开运算 2.1 代码示例 2.2 运行结果 3. 开运算接口 3.1 参数详解 3.2 代码示例 3.3 运行结果 4. 开运算应用场景 5. 注意事项 6. 总结 相关阅读 OpenCV:图像的腐蚀与膨胀-CSDN博客 OpenCV:闭运算-CSDN博客 …...

38. RTC实验
一、RTC原理详解 1、6U内部自带到了一个RTC外设,确切的说是SRTC。6U和6ULL的RTC内容在SNVS章节。6U的RTC分为LP和HP。LP叫做SRTC,HP是RTC,但是HP的RTC掉电以后数据就丢失了,即使用了纽扣电池也没用。所以必须要使用LP,…...

Flutter 新春第一弹,Dart 宏功能推进暂停,后续专注定制数据处理支持
在去年春节,Flutter 官方发布了宏(Macros)编程的原型支持, 同年的 5 月份在 Google I/O 发布的 Dart 3.4 宣布了宏的实验性支持,但是对于 Dart 内部来说,从启动宏编程实验开始已经过去了几年,但…...

巴菲特价值投资思想的核心原则
巴菲特价值投资思想的核心原则 关键词:安全边际、长期投资、内在价值、管理团队、经济护城河、简单透明 摘要:本文深入探讨了巴菲特价值投资思想的核心原则,包括安全边际、长期投资、企业内在价值、优秀管理团队、经济护城河和简单透明的业务…...

从零构建可视化爬虫管理平台:ClawPanel架构设计与实战
1. 项目概述与核心价值最近在折腾一个自动化数据采集的小项目,偶然在GitHub上看到了一个名为“ClawPanel”的开源项目,作者是zhaoxinyi02。这个项目名字直译过来是“抓取面板”,光看标题就让我这个老爬虫工程师眼前一亮。在数据驱动的今天&am…...

AI编程技能自学习:构建Claude与Cursor的智能协同开发环境
1. 项目概述:当Claude遇上Cursor,一场关于AI编程技能的自我进化最近在GitHub上看到一个挺有意思的项目,叫Self-Learning-Claude-Skill。虽然项目描述和正文都还是空的,但光看这个标题和关键词——claude-code、cursor、skills——…...

AI研究代理基准测试工具autoresearch-adal:自动化对比AdaL与Claude Code
1. 项目概述与核心价值如果你和我一样,经常在多个AI研究工具之间切换,试图找出哪个模型在解决复杂的、需要多步推理的研究任务上更胜一筹,那么你肯定体会过那种繁琐和低效。手动设置不同的API环境、编写重复的测试脚本、整理散落在各处的输出…...

Seelen UI定制化桌面
链接:https://pan.quark.cn/s/0d0312d1a6d1Seelen UI是适用于 Windows 10/11的第一个基于 Web 的完全可定制的桌面环境,提供了一种直观而强大的方式来管理和自定义您的工作区。提升工作效率与体验,满足不同用户的需求。...

闲置烽火HG680L变身全能播放器:S905L-3B芯片刷机后安装EmuELEC游戏系统+CoreELEC影音库
闲置烽火HG680L改造指南:打造全能家庭娱乐终端 家里角落积灰的烽火HG680L机顶盒,其实是一块被低估的硬件宝藏。搭载Amlogic S905L-3B芯片的它,性能远超普通电视盒子。通过巧妙改造,不仅能流畅运行复古游戏系统,还能变身…...
)
别再手动画图了!用Python ASE + Matplotlib一键生成高质量材料结构图(附完整代码)
科研绘图革命:用Python ASEMatplotlib实现材料结构可视化自动化 深夜的实验室里,屏幕荧光映照着一张疲惫的脸——这可能是许多材料科学研究者共同的记忆。当你在论文截稿日前夕,还在反复调整VESTA中的原子位置、尝试各种角度截图时࿰…...

创业沟通陷阱:从“一切顺利”到“坦诚求助”的工程化实践
1. 项目概述:当“独角兽”闭上嘴,“彩虹”褪了色在科技创业圈混了十几年,从硅谷到深圳,从硬件孵化器到软件路演日,我见过太多这样的场景。你走进一个挤满创业者的房间,空气里弥漫着咖啡因和焦虑混合的独特气…...

扰动补偿自触发MPC控制器设计【附代码】
✨ 长期致力于永磁同步电机、模型预测控制、扰动补偿、死区时间优化、自触发控制研究工作,擅长数据搜集与处理、建模仿真、程序编写、仿真设计。 ✅ 专业定制毕设、代码 ✅ 如需沟通交流,点击《获取方式》 (1)基于预测误差驱动的扰…...
)
用ChatGPT 10分钟生成TikTok爆款脚本:5步工作流+3类高转化话术模板(附Prompt库下载)
更多请点击: https://intelliparadigm.com 第一章:ChatGPT TikTok视频创意 在短视频爆发式增长的今天,TikTok 内容创作者亟需高效、可复用的创意生成机制。ChatGPT 可作为智能脚本引擎,将抽象主题快速转化为结构化、高传播性的视…...

从丰田SUA事件看安全关键系统软件可靠性:设计原则与工程实践
1. 项目概述:当软件缺陷成为致命威胁我干了十多年嵌入式开发,从单片机玩到复杂的汽车域控制器,经手的代码行数自己都数不清了。但每次看到“软件缺陷导致车辆突然加速”这类新闻,后背还是会发凉。这行干久了,你会对代码…...