GRN前沿:GRETA:从多模式单细胞数据推断基因调控网络方法的比较与评价
1.论文原名:Comparison and evaluation of methods to infer gene regulatory networks frommultimodal single-cell data
2.发表日期:20254.12.21

摘要:
细胞通过基因表达调节其功能,由转录因子和其他调节机制的复杂相互作用驱动,这些机制一起可以被建模为基因调节网络(GRNs)。单细胞多组学技术的出现推动了整合转录组学和染色质可及性数据以推断GRNs的几种方法的发展。虽然这些方法提供了它们在发现新的监管相互作用方面的实用性的例子,但缺乏评估其机制和预测特性以及恢复已知相互作用的能力的全面基准。为了解决这个问题,我们建立了一个全面的框架,基因调控网络分析(GRETA),可作为Snakemake管道,其中包括以模块化方式分解其不同步骤的最先进方法。有了它,我们发现GRN对方法的选择非常敏感,例如随机种子的变化,或替换推理管道中的步骤,以及它们是否使用配对或未配对的多模态数据。虽然获得的网络在预测评估任务中表现良好,并部分恢复了已知的相互作用,但它们很难从扰动分析中捕获因果关系。我们的工作引起了人们对从单细胞组学推断GRNs的挑战的关注,提供了指导方针,并为开发和测试新方法提供了灵活的框架。
关键词:基因调控网络,基因调控,方法,基准,多组学,单细胞
GRETA框架
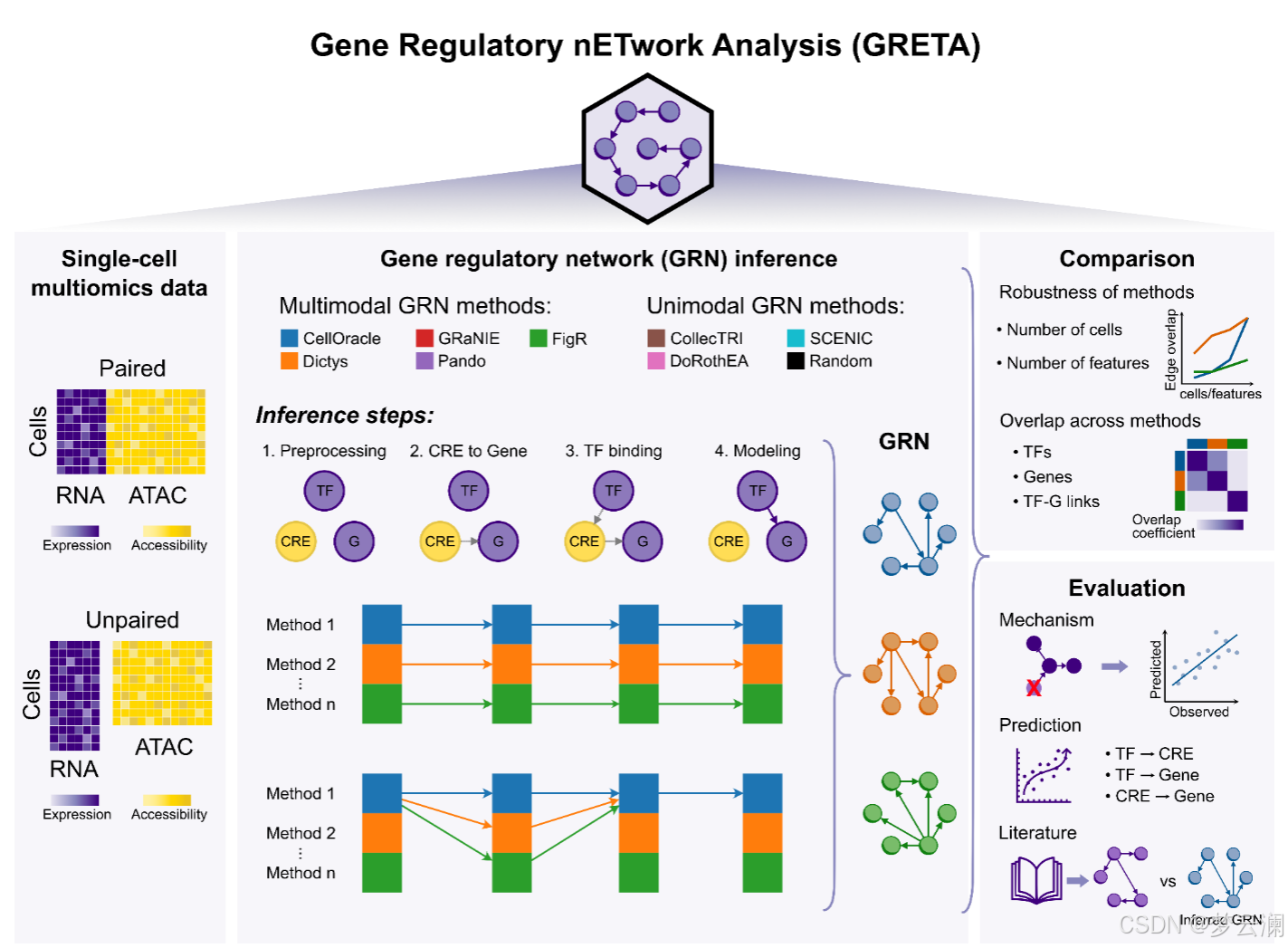

GRETA能够在转录因子(TF)、边和目标基因层面对基因调控网络(GRNs)进行系统的比较。为了考虑网络大小的差异,它使用了重叠系数,该系数定义为两个集合之间的交集大小除以较小集合的大小。利用这一统计量,它还可以通过在推断过程中使用不同的随机种子,或者通过降采样特征或细胞来测量方法的稳健性。
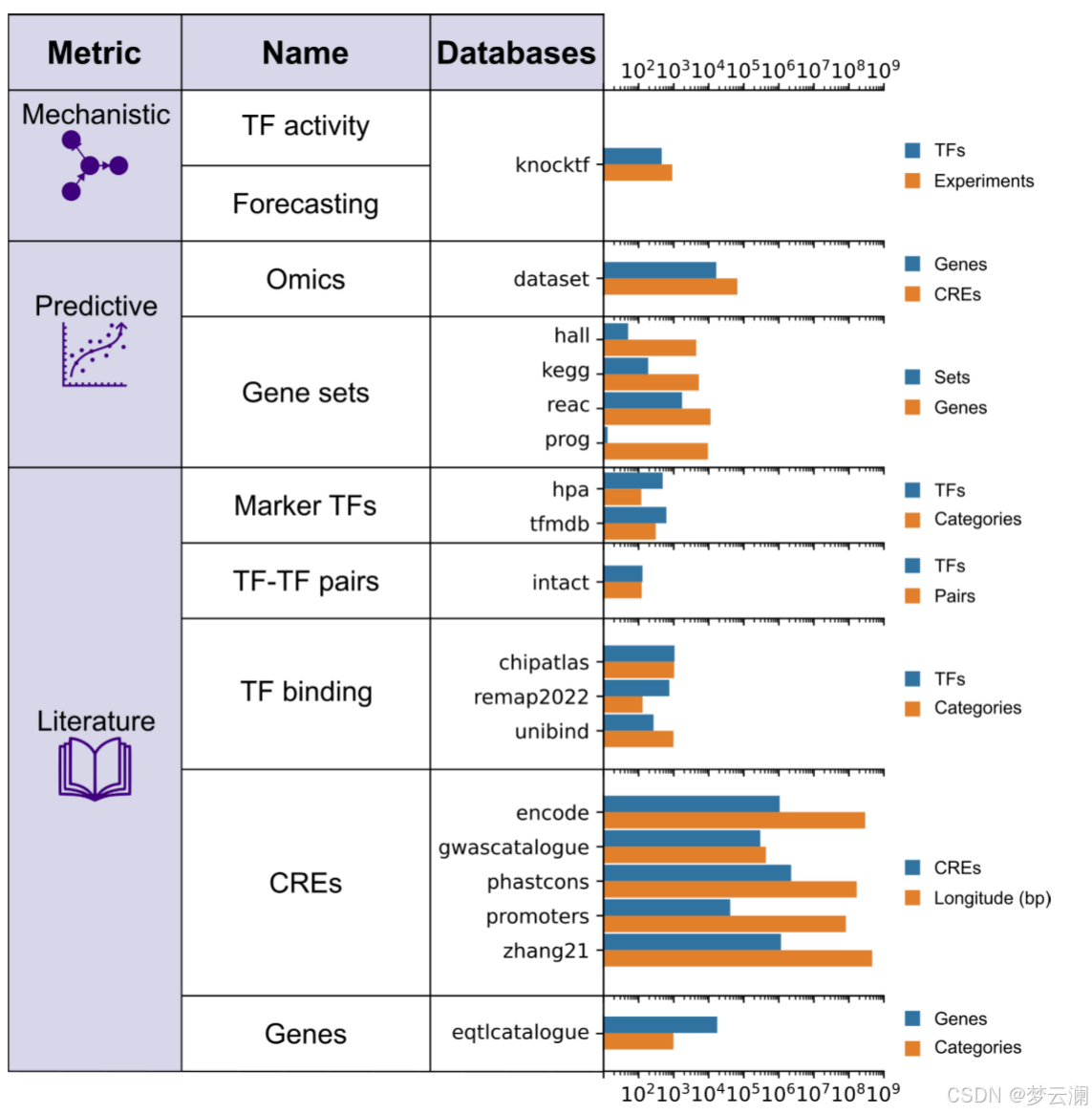
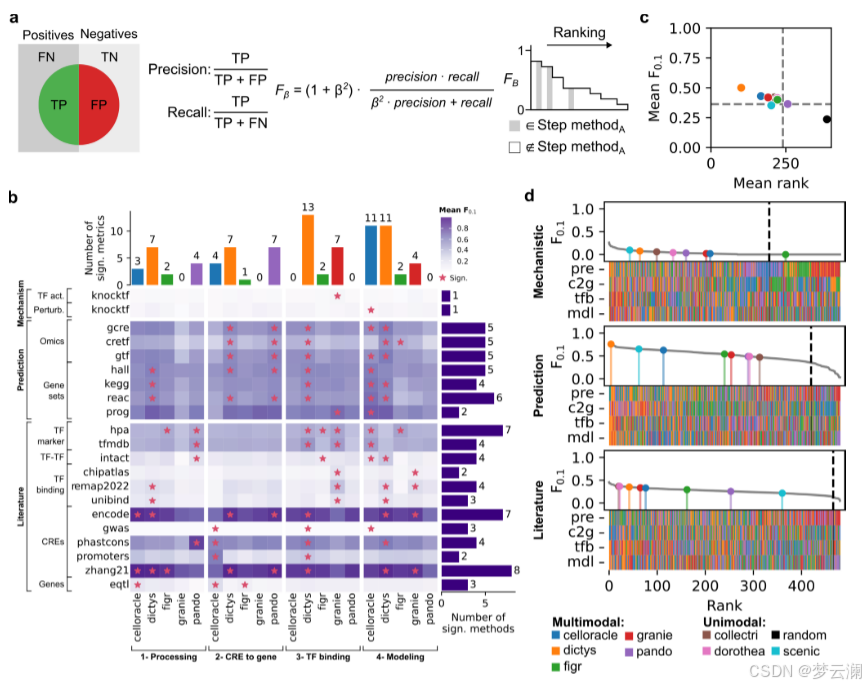
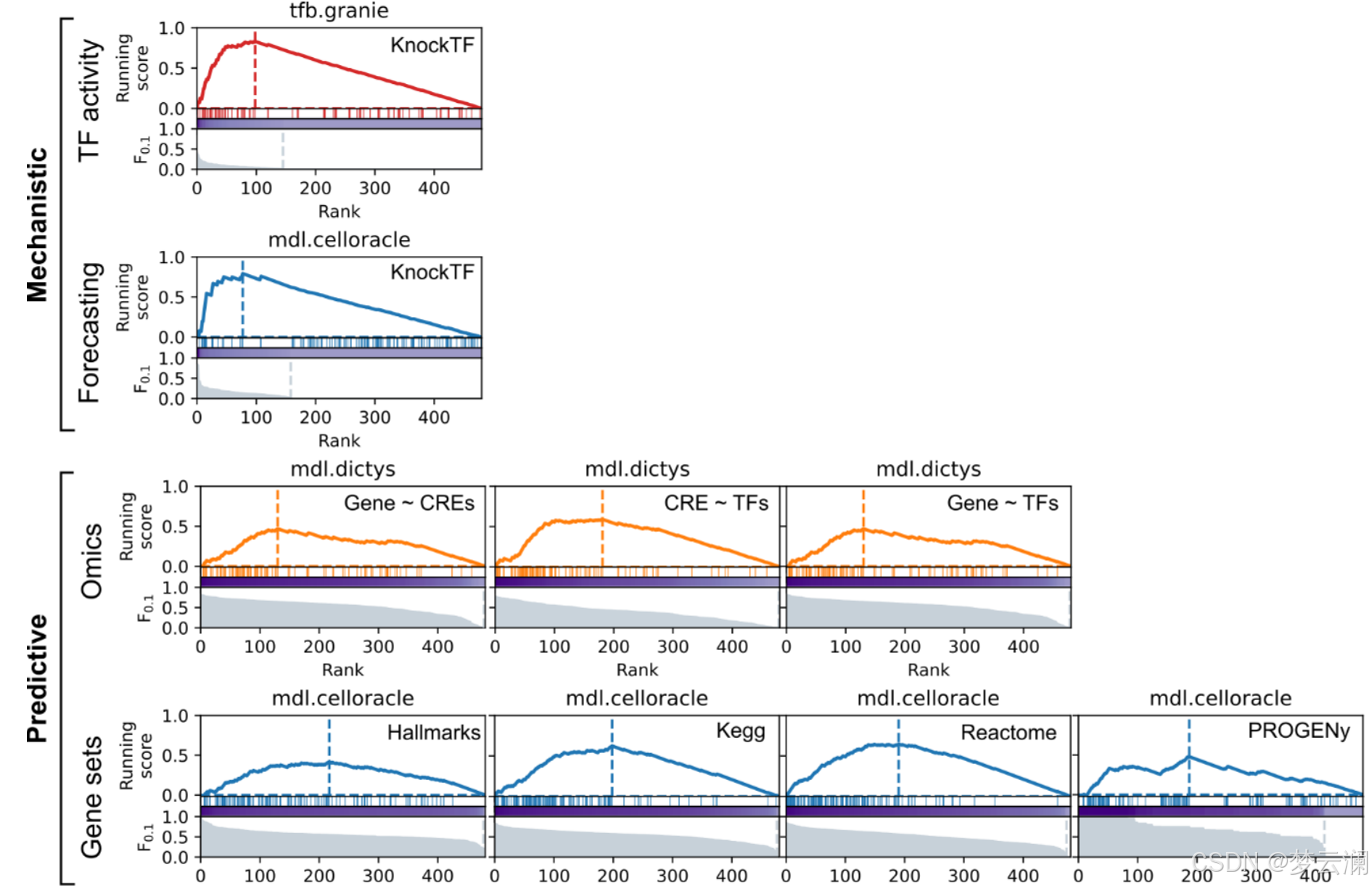
为了扩展基因调控网络(GRN)的基准测试策略,超越通过ChIP-seq数据测量的转录因子(TF)结合峰的恢复,并结合具有远端顺式调控元件(CREs)的新型多模态GRNs,GRETA提供了一组多样化的指标,分为三类:机制性、预测性和基于文献的指标(图2)。每个指标的详细目标和评估程序在表1和方法部分提供。
机制性指标评估GRNs是否能够通过评估两个方面来捕捉扰动实验中的因果关系:它们使用基因签名的富集分数识别受扰动的TFs的能力(“TF活性”)以及它们在预测扰动后基因表达变化的准确性(“预测”)(方法)。
预测性指标评估GRN拓扑结构如何模拟观察到的单细胞RNA-seq和ATAC-seq数据(“组学”)和基因集富集分数(“基因集”)(方法)。
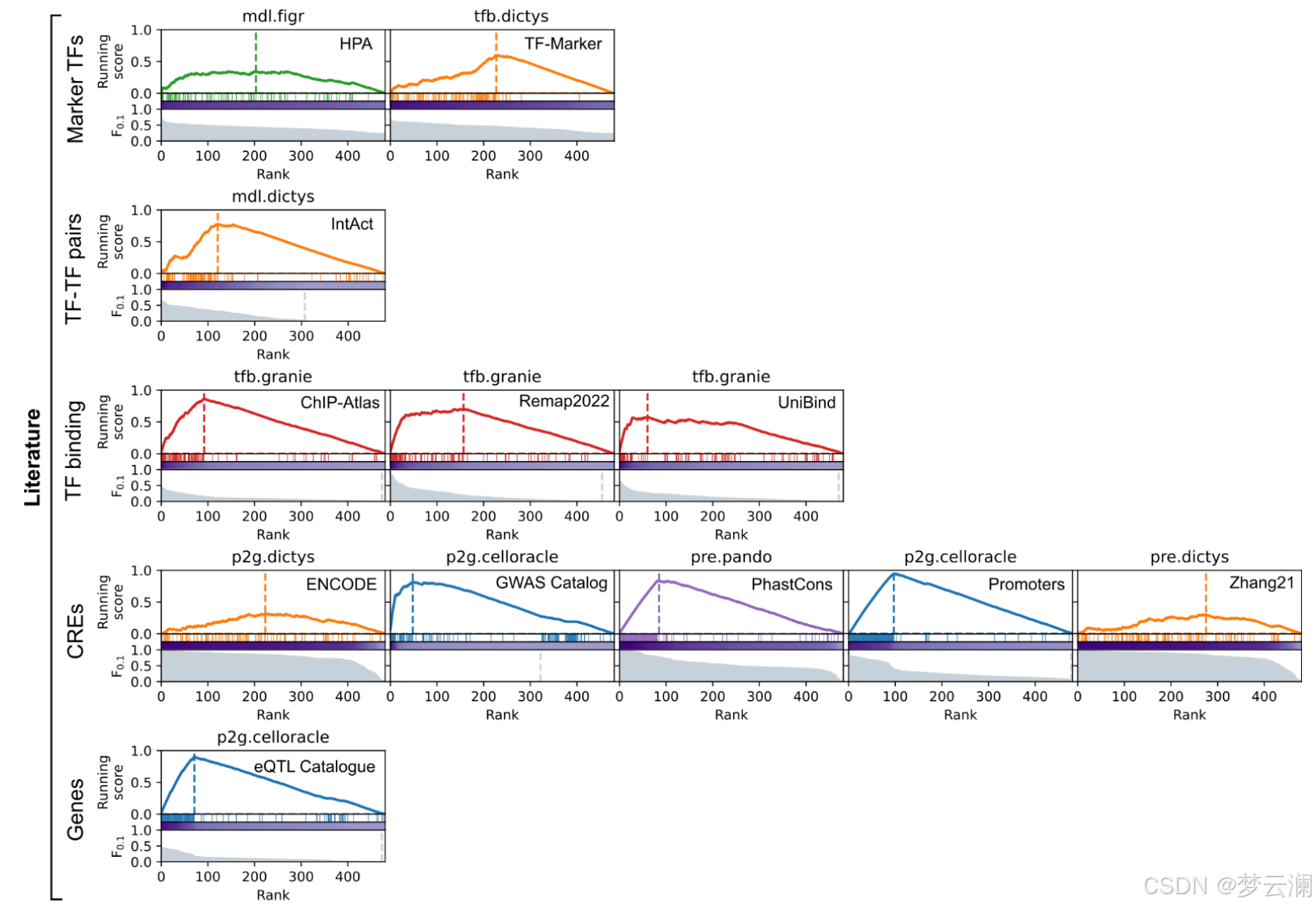
基于文献的指标衡量GRNs在多大程度上结合了来自数据库的先验知识,关注几个方面:包括已知的TF标记(“标记TFs”);基于共享目标基因的TF-TF蛋白-蛋白相互作用(“TF-TF对”);通过独立ChIP-seq数据验证的TF结合事件到CREs(“TF结合”);通过生化实验、进化保守性或遗传变异存在性识别为功能活跃的CREs;以及通过表达数量性状位点(eQTLs)与基因相关联的CREs(方法)。
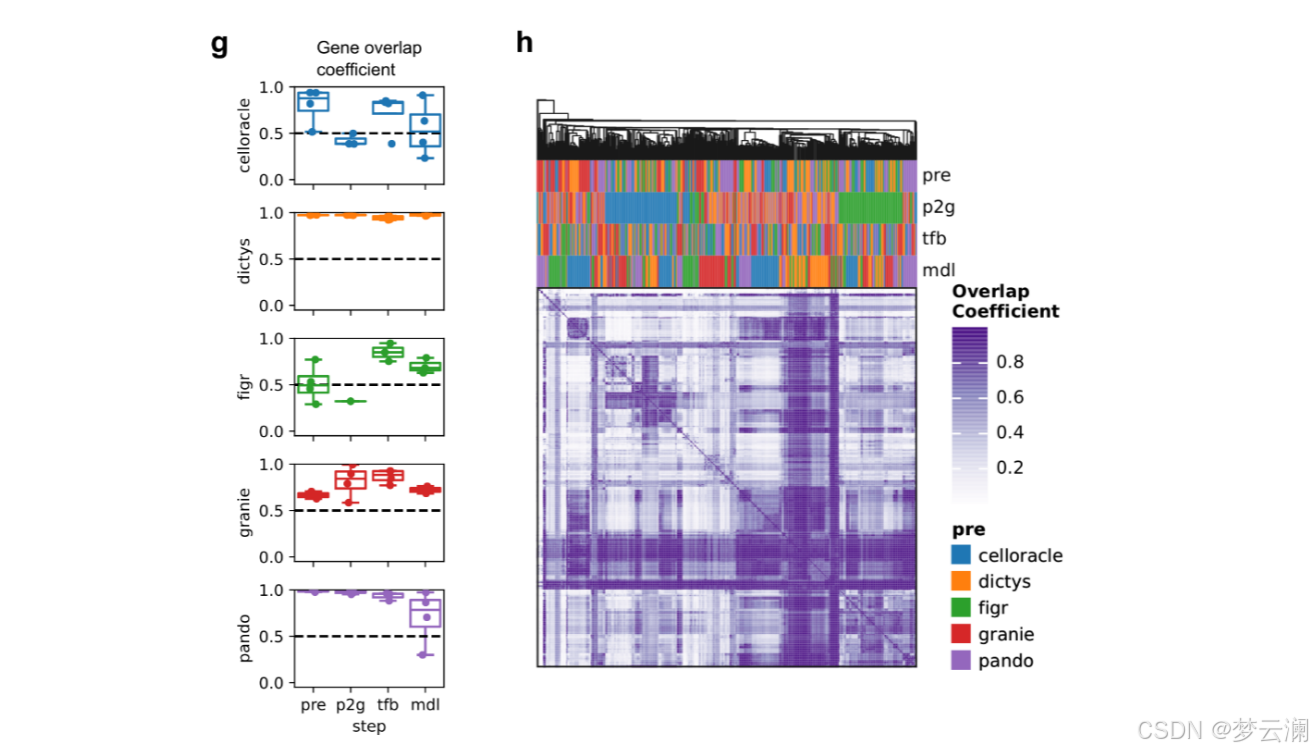
在处理完所有评估数据库后,我们观察到目标基因的覆盖率很高,转录因子(TFs)的覆盖率适中,而跨资源的基因组区域的覆盖率较低(平均重叠系数基因=0.7528;平均重叠系数TFs=0.5382;平均重叠系数碱基对=0.311)(扩展数据图1a-c)。
![]()
1.多种方法推导的GRNs的系统比较和稳定性定量
我们使用GRETA重建了一个公开可用的、经过注释的人类垂体单细胞多组学数据集的基因调控网络(GRN),使用了五种多模态GRN重建方法、SCENIC、两个先验知识GRNs和随机GRN的原始实现(方法)。
首先,我们评估了这些方法在重现GRN推断中的稳健性,使用相同的垂体数据集进行了三次不同的随机种子运行。大多数方法在每次运行中都能一致地恢复相同的转录因子(TFs)和目标基因(平均重叠系数TFs = 0.96;平均重叠系数基因 = 0.95),以及它们的相互作用(平均重叠系数边 = 0.85)(扩展数据图2a)。然而,Dictys和SCENIC在边的重叠上显示出显著减少,重叠系数值低于0.5(图3a)。接下来,我们评估了那些在随机种子间未达到完全重叠(重叠系数 = 1)的方法在重复实验中的交互权重的一致性。大多数方法显示出强烈的相关性,反映了稳定的交互推断(中位Pearson相关系数ρ = 0.96)(扩展数据图2b)。
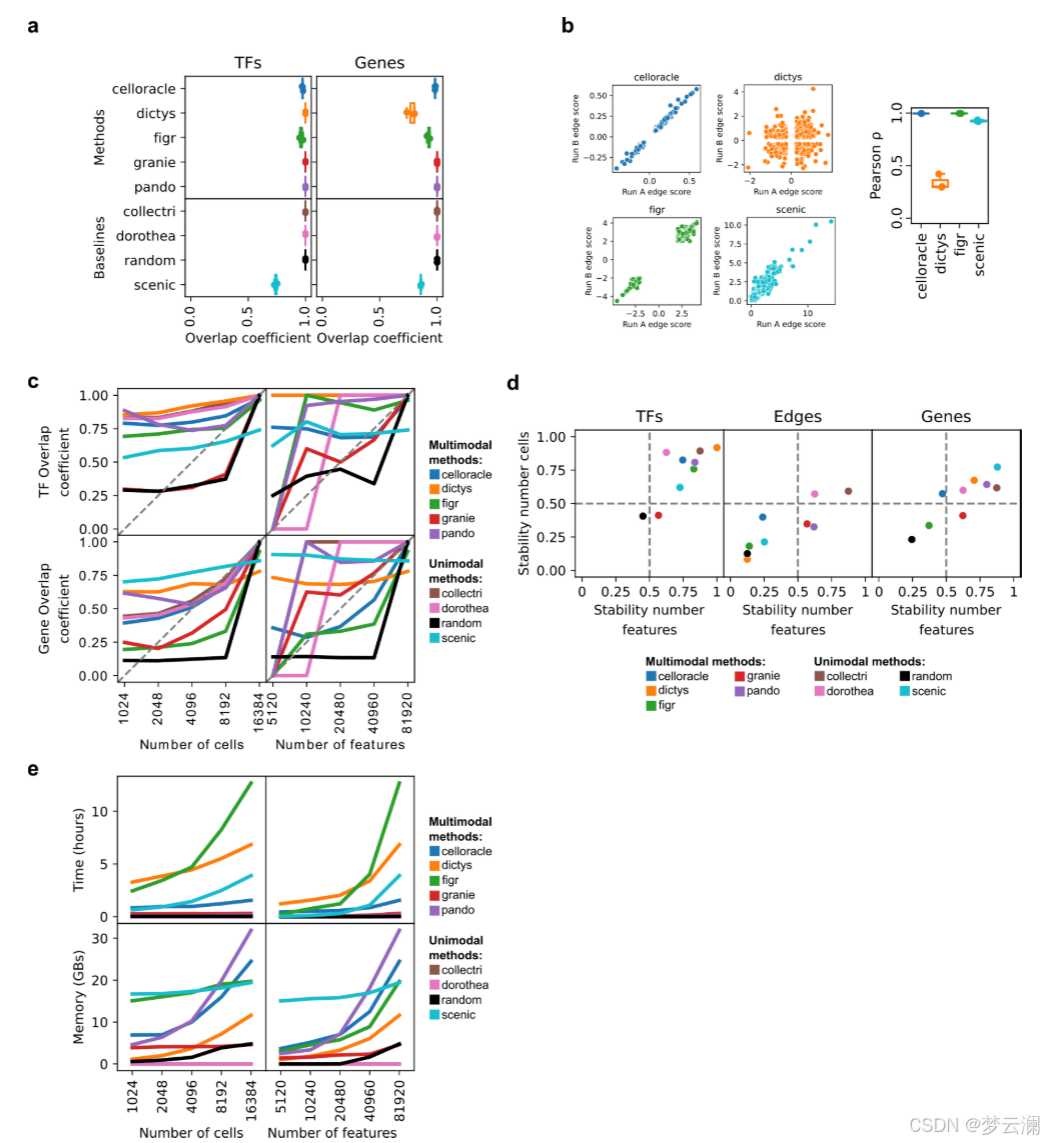

然而,Dictys显示出低于0.5的相关值,并且在运行之间高度加权的TF-基因交互方向发生了反转。这些发现表明,某些方法的概率性质可能会影响GRN推断并影响下游分析。
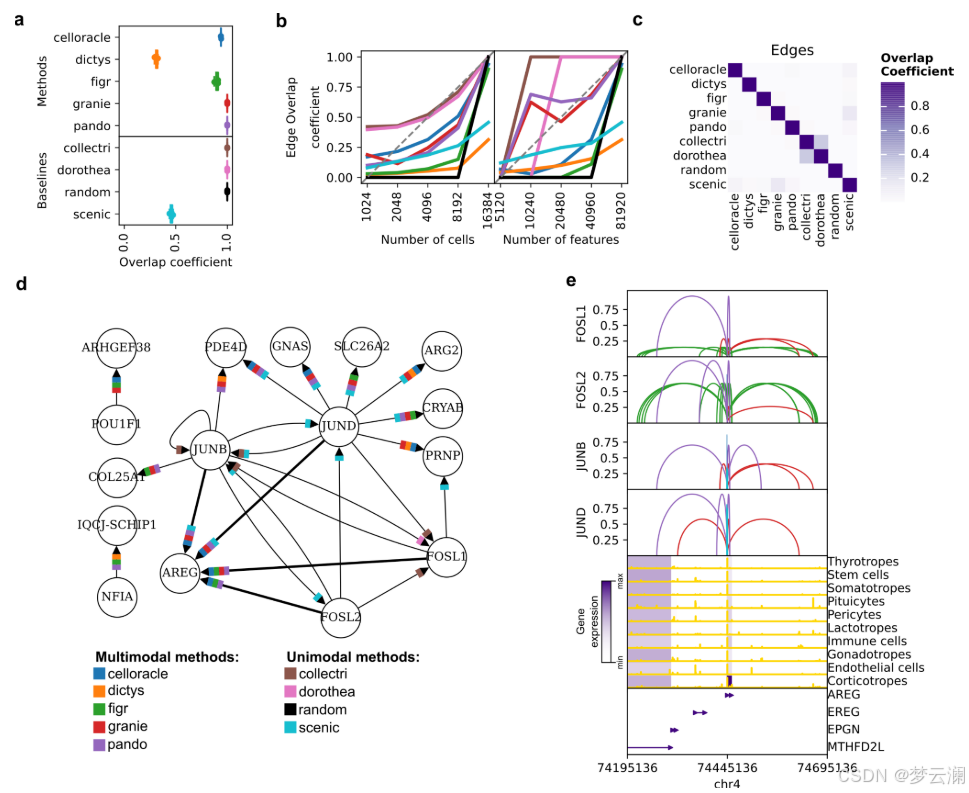

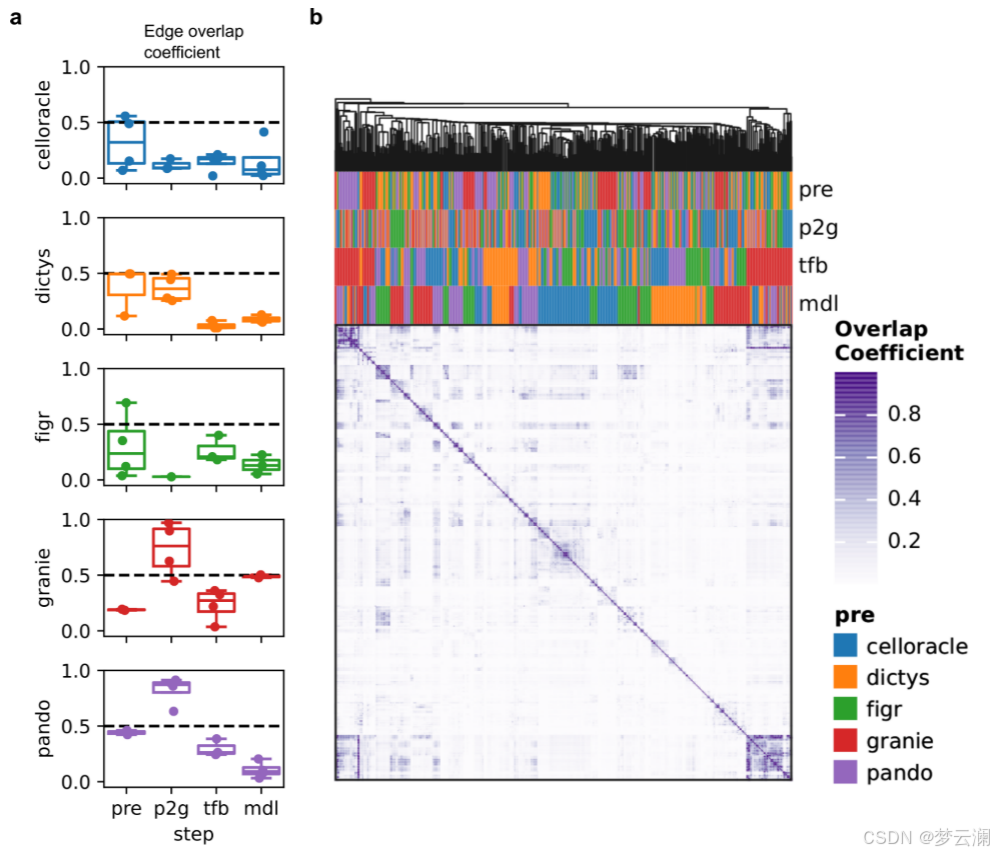
我们接下来研究了数据规模如何影响GRN重建,因为从同一采样组织中保留的细胞或特征数量可能会因实验方案和单细胞数据处理框架及其选择的参数而异。垂体数据集在特征或细胞数量上逐渐降采样,分为四个递减的值,并且如前所述推断GRN。对于每个降采样级别,使用了三个不同的随机种子。结果网络与使用完整数据集生成的网络在TF、边和目标基因层面上使用重叠系数进行比较。基于重叠系数曲线下的面积计算稳定性分数,接近1的值表示对降采样的影响最小,接近0的值表示影响较大(方法)。减少细胞和特征的数量并没有显著影响TF和基因的恢复,重叠系数有适度的减少(TF和基因的平均稳定性分数为0.73)(扩展数据图2c,d)。然而,我们观察到所有方法在边层面上的相似性急剧下降(平均稳定性分数为0.35)(图3b,扩展数据图2d)。在降采样策略期间对运行时间和内存使用的测量强调了多模态方法对计算资源的高要求(补充说明1,扩展数据图2e)。这些发现表明,数据覆盖强烈影响网络结构,尤其是相互作用,强调需要优化预处理以保留关键网络特征。
接下来,我们比较了使用不同方法从同一数据集中获得的基因调控网络(GRNs)。大多数方法推断出的GRNs具有相似数量的转录因子(TFs)、边和每个TF的目标基因数量(中位数TFs = 245,边 = 2,663,基因 = 1,569,调控子大小 = 17.3)(扩展数据图3a)。然而,Dictys和SCENIC产生了更大的网络,拥有超过20,000个边,近10,000个目标基因,以及超过50个基因的调控子。此外,尽管所有方法都被设置为使用相同的距离参数(距离转录起始位点TSS ± 250 kb),但方法将基因与位于不同基因组距离的顺式调控元件(CREs)联系起来(中位数CRE到TSS的距离 = 12.7 kb)(扩展数据图3b)。特别是,CellOracle主要只恢复了靠近TSS的启动子区域,而Pando有时会识别超出距离限制的远端相互作用。总之,不同方法获得的GRNs在拓扑结构上有显著差异。
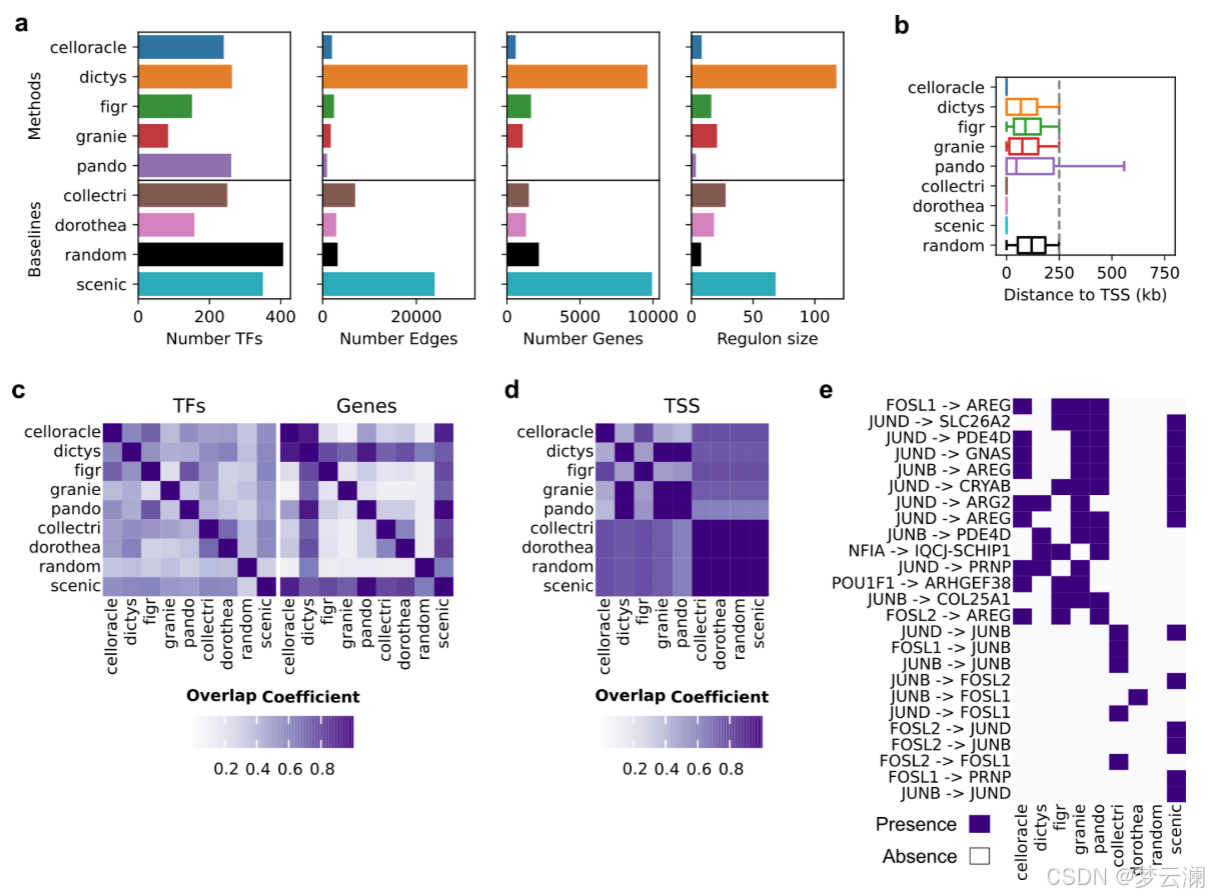

在比较不同方法获得的GRNs的重叠时,它们在TF和目标基因层面上显示出适度的一致性(平均重叠系数TFs = 0.46;平均重叠系数基因 = 0.45)(扩展数据图3c),但在边层面上的重叠最小(平均重叠系数边 = 0.02)(图3c)。我们还发现不同方法中使用的不同的TSS注释之间存在显著差异(平均基因组重叠系数 = 0.7692)(扩展数据图3d)。特别是,CellOracle和FigR的注释与其他注释的重叠最少(平均基因组重叠系数CellOracle = 0.6377,FigR = 0.66)。虽然其他方法使用了Ensembl的hg38基因注释版本的相似版本,但CellOracle使用了使用Homer推断的自定义注释,而FigR使用了hg19 RefSeq。这些结果表明,尽管这些方法共享相似的推断步骤,但它们的算法选择和先验知识的使用影响了GRN重建。
尽管这些方法在边层面上显示出有限的重叠,但对于涉及转录因子FOSL1、FOSL2、JUNB和JUND等的小型调控网络,每种调控相互作用至少在五种多模态方法中的三种中是反复出现的(图3d,扩展数据图3e)。GRN方法共同识别出AREG,这是一种表皮生长因子受体的信号配体,作为这些转录因子高度调控的目标基因。AREG既不存在于文献衍生的GRNs中,也不存在于随机网络中。虽然SCENIC捕获了其中一些相互作用,但它推断出的边的数量比多模态GRN方法要多,这可能是由于它完全依赖于转录组学,这可能会增加假阳性的可能性。这些结果说明了GRN推断方法如何揭示以前未被描述的调控相互作用。
为了理解不同方法如何证明与AREG的调控联系,我们在基因组上可视化了它们预测的TF-CRE-基因相互作用(图3e)。虽然AREG的启动子区域在所有细胞类型中都是可接近的,但其表达仅限于促肾上腺皮质激素细胞,这是一种垂体细胞类型,它在下丘脑刺激下分泌应激激素。这表明AREG的表达受到特定上下文的远端顺式调控相互作用的影响。
总的来说,本节的结果突出了网络规模和连接性在不同方法之间的可变性,反映了它们在推断步骤和算法策略上的差异。
2.评估配对与非配对多组数据在GRN推理中的影响
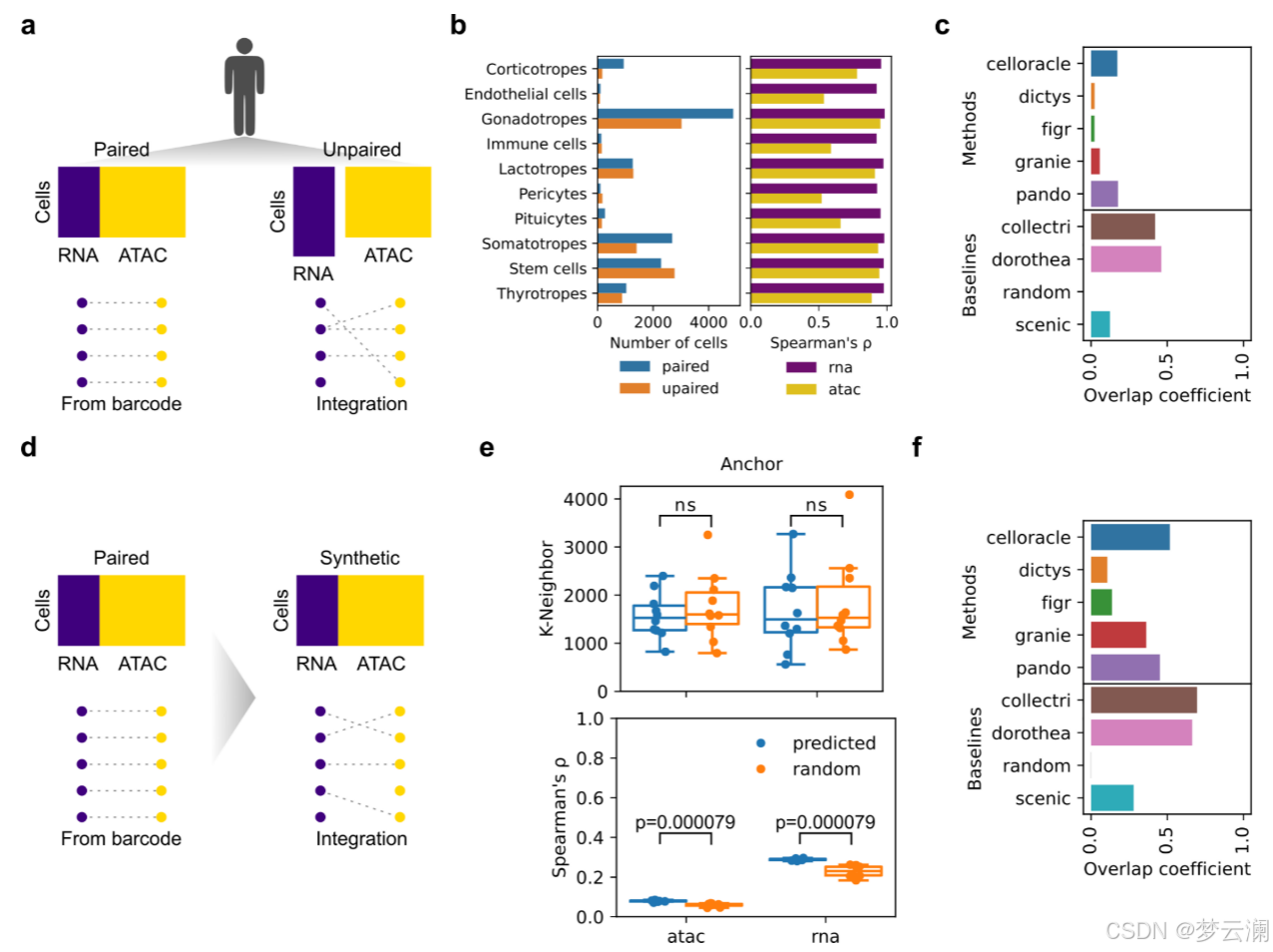
在进行基因调控网络(GRN)推断时,需要考虑的另一个关键因素是所使用的多模态数据的性质。数据是否真正成对,即同时测量来自相同细胞的单细胞RNA测序(snRNA-seq)和单细胞ATAC测序(snATAC-seq),或者是不成对的,即从不同细胞测量,这可能会影响GRN重建。虽然一些方法,如CellOracle和GRaNIE,被设计为能够处理成对和不成对的数据,但其他方法仅限于成对数据,并依赖于计算策略来整合在处理不成对数据集时的模态。为了评估这些差异对GRN推断的影响,我们再次使用了人类垂体数据集,该数据集独特地包含了使用成对和不成对多组学技术生成的同一供体的资料,使其特别适合进行这种比较。

然后,我们比较了每种方法从两个数据集中推断出的GRNs,并发现网络边缘之间的重叠度很低(平均重叠系数边缘 = 0.14)。这些结果表明,虽然成对和不成对的多组学数据集保留了全局细胞类型属性,但不成对数据的计算整合引入了影响推断GRNs的差异。



3.分解多模态GRN推理
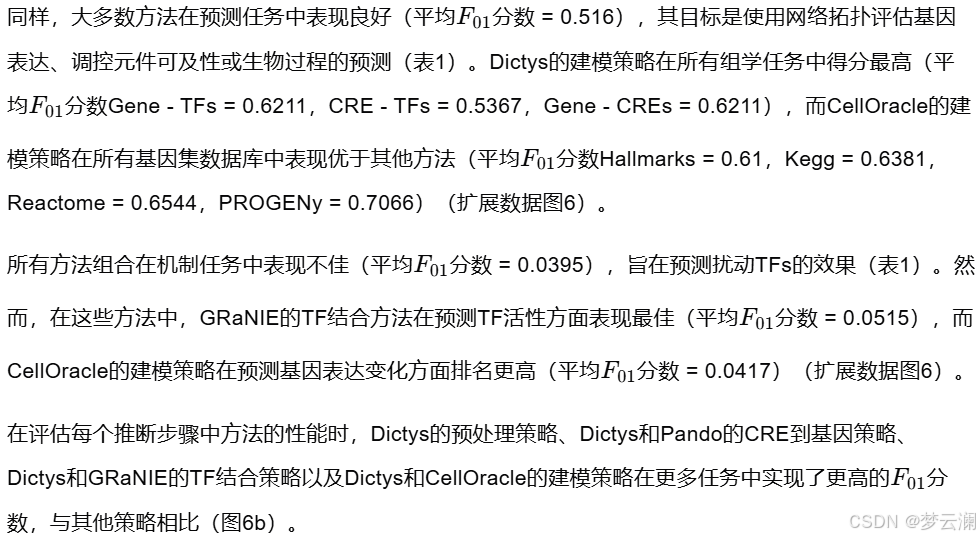
为了评估多模态方法中每个推断步骤在GRNs重建中的影响,我们使用了外周血单个核细胞的成对多组学数据集(扩展数据图5a-c),并系统地将每个原始方法中的个别步骤替换为其他方法中的步骤(方法)。为了验证我们的实现,我们将GRETA的多模态GRN方法的解耦版本与原始版本进行了比较。除了Dictys在其原始实现中显示出GRN推断的不稳定性(结果2.2)外,所有其他方法都表现出高度的一致性,具有显著的边重叠系数和原始与GRETA实现之间的Spearman相关系数(平均边重叠系数 = 0.78;平均Spearman相关系数 = 0.85)(扩展数据图5d)。
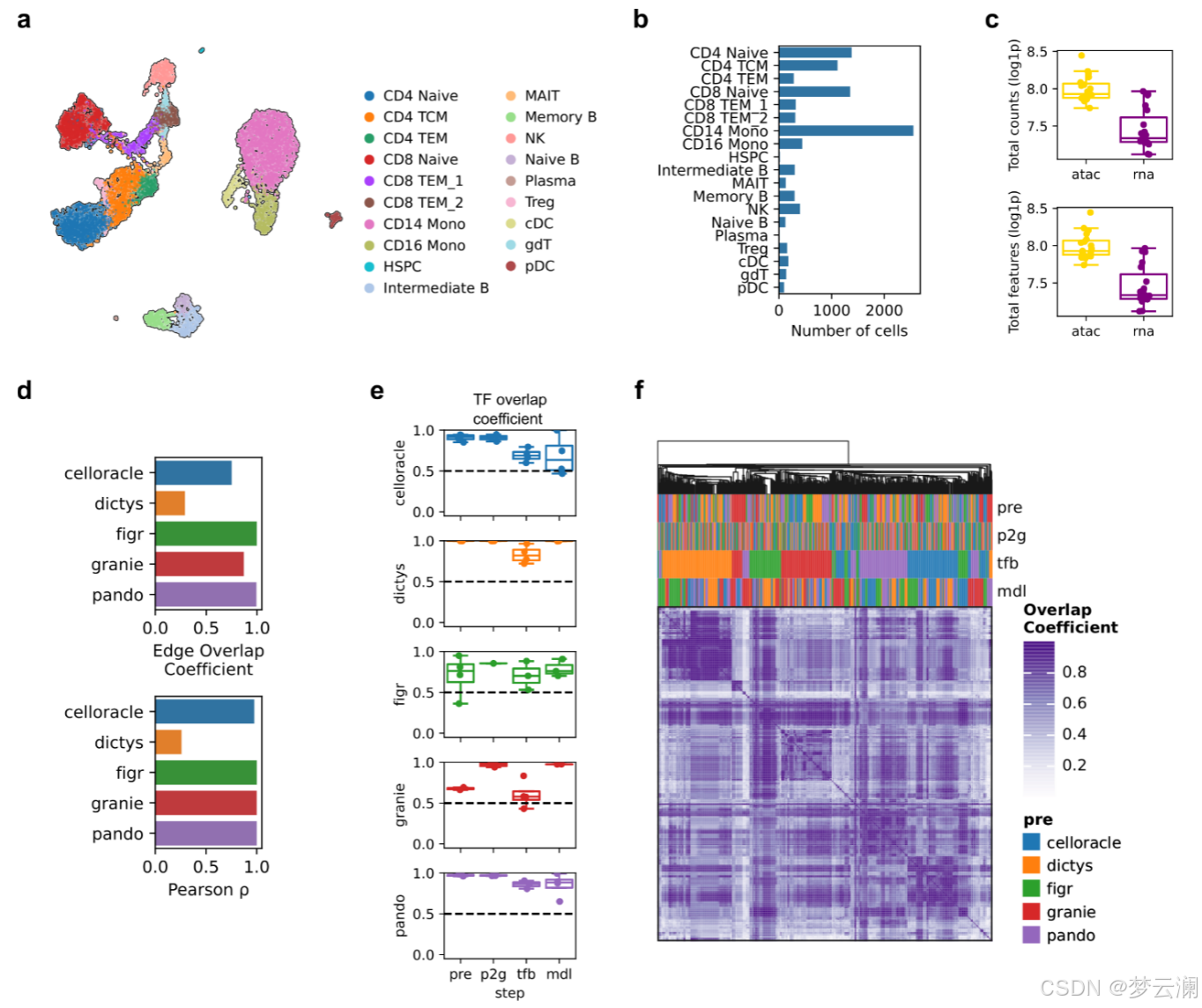

接下来,我们跨方法组合推断步骤,并观察到用其他方法的步骤替换个别步骤对GRN重建有不同的影响。在不同的组合中,包含在GRNs中的TFs和基因的重叠系数始终保持在0.5以上,尽管基因级别的重叠显示出更大的变异性(TF平均重叠系数 = 0.8461;基因平均重叠系数 = 0.7901;扩展数据图5e-h)。然而,在边的层面上,修改单个分析步骤会显著改变GRN结构,导致重叠系数降低(平均重叠系数 = 0.2877;图5a)。
![]()

4.多模态GRNs的评价




![]()
方法
1.多组数据处理
从GEO、CELLxGENE和10x Genomics门户为每个数据集检索了原始转录计数矩阵、样本特异性染色质可及性片段文件和全局条形码注释。片段文件被处理以将相应的样本名称分配给条形码,确保它们是唯一的。每个数据集都独立地使用下面描述的相同处理流程进行处理。
对于数据集中的每个样本,片段文件使用SnapATAC2处理成瓦片矩阵。删除了不在每个数据集注释元数据中的条形码,保证只保留高质量的条形码。瓦片矩阵被连接起来,并使用SnapATAC2实现的MACS8调用数据集特定的共识可及性峰(顺式调控元件,CREs)。
在转录计数矩阵中移除了在少数细胞中表达的基因,以及包含少量表达基因的细胞(基因数量 > 100,细胞数量 > 3)。如果转录计数矩阵中的基因符号不在Ensembl 111中,则将其移除。对于每个数据集,如果一个基因符号出现多次,则保留检测到最多细胞数量的特征。只有在相应数据集注释中存在的条形码才会被保留。然后,转录和可及性矩阵被配对成一个MuData对象。

2.snRNA-seq和snATAC-seq配对
为了整合不成对的snRNA-seq和snATAC-seq垂体数据集,我们首先使用Seurat生成了一个共享的共嵌入。对于RNA数据,我们执行了对数标准化,选择了高变异基因,并缩放了数据。对于ATAC数据,我们应用了词频-逆文档频率(TF-IDF)标准化,选择了高变异的CREs,并同样进行了缩放。从ATAC数据中推断出基因分数,然后将其作为RNA进行后续处理。然后使用典型相关分析来推导出RNA和推断出的基因分数之间的共享嵌入。使用FigR基于最优传输的整合方法,将单个细胞以一对一的方式进行配对。最后,将匹配的条形码合并成一个成对的MuData对象。相同的程序用于从真实的成对垂体数据集中生成合成的成对数据集。
3.原始多模态GRN推理方法实现



4.稳定性得分
对于特征和细胞,计算了TFs、边和基因重叠系数在逐渐减少的降采样迭代过程中的稳定性得分。这个得分是为每种方法生成的,通过计算重叠系数曲线下的面积(AUC)来实现。AUC是通过将梯形规则应用于从0到1的归一化降采样步骤序列来确定的,该序列有五个步骤,并对应于该方法的重叠系数值。这种方法提供了一种定量衡量GRN方法在通过细胞或特征数量降采样时稳定性的方法。
5.单峰GRN推理方法
本节描述了那些不直接对多模态数据进行建模,但仍然可以通过先验知识整合CRE信息的GRN方法。文献衍生的GRNs,CollecTRI和DoRothEA,分别从它们的Zenodo和GitHub存储库中获取。对于每个数据集,从GRNs中移除了非表达基因。然后,为每个基因添加了一个CRE,包括其启动子区域(从Ensembl 111定义的TSS起±1kb)。最后,仅当TF调节子包含超过五个目标基因时才保留。
SCENIC首先使用该方法提供的自定义注释将基因分类为TFs或非TFs。对于每个基因,它然后拟合了一个XGBoost模型,以基于TF基因的表达来预测其观察到的表达。接下来,基于TF基序结合在它们启动子区域的富集,修剪每个TF调节子内的目标基因。最后,通过分数过滤相互作用,仅保留那些分数>0.001的。
对于每个数据集,通过随机采样25%的表达基因生成了一个随机GRN。对于每个采样基因,基于Ensembl 111的TSS注释,使用指数分布(比例=1)在500 kb窗口内选择邻近的可及CREs。对于每个采样的CRE,从Lambert的数据库中采样注释的TFs,这些TFs也存在于采样的基因中,使用指数分布(比例=1)进行类似采样。结果元素被合并,TF调节子被随机子采样以实现10%的TF到基因的比例。
6.解耦多模态GRN推理方法
对于每种原始方法,我们实现了一个解耦版本,旨在与其他推断步骤兼容。虽然这些单独的步骤在很大程度上反映了原始实现,但在一些方法中进行了小的修改,以确保不同方法组合的最大兼容性。
对于CellOracle,基于UMAP坐标中邻近性的可及性插补全在与Cicero进行CRE-CRE交互推断时没有进行,因为在UMAP空间中的距离没有意义。
对于Dictys,使用替代策略预处理的数据集也结合了Dictys的方法来减少潜在的稀疏性。此外,在建模之前构建脚手架GRN时,仅包括在CRE-基因关联步骤中识别的峰值。
对于GRaNIE,首先执行CRE到基因的关联,然后在这些关联的CREs中进行TF结合预测,而不是所有可用的CREs。采用这种方法是因为在原始实现中,许多具有TF结合预测的峰值最终在CRE到基因合并步骤中被排除。此外,解耦的流程需要一个顺序处理顺序。
同样,对于Pando,处理顺序从原始实现中进行了调整:首先执行CRE-基因关联,然后进行TF结合步骤。此外,为了确保与FigR的兼容性,在MOODS中获得的负对数(p值)在罕见的情况下被设置为零。
7.评价指标
对于机制指标,从KnockTF中检索了单转录因子(TF)扰动的对数变化及其相关元数据。对于“TF活性”指标,基于与正在评估的数据集相关的术语对扰动实验进行筛选。如果扰动的TF存在于GRN中,则使用decoupler-py的ULM方法推断TF富集分数。如果获得的富集分数既是正的又显著(BH FDR < 0.05),则认为扰动是一个真正的阳性(TP)。
对于“预测”指标,通过匹配术语对实验进行类似的筛选。当TF存在于GRN中时,扰动的影响通过首先平均原始单核多组学数据集中所有细胞的基因表达值,然后使用CellOracle的扰动框架(步骤数=3)进行模拟扰动模拟来模拟。当模拟变化与测量变化之间的Spearman相关性显著(p > 0.05;BH FDR < 0.05)时,扰动被认为是TP。
对于预测指标“Omics”,原始单核多组学数据集中的细胞按细胞类型分层、采样,并分为训练和测试集(训练集大小 = 66%,测试集大小 = 33%)。然后使用XGBoost来建模以下关系:基因 ~ TFs,基因 ~ CREs,以及CREs ~ TFs。基因和TFs使用对数标准化基因表达,而CREs使用对数标准化染色质可及性。在拟合模型后,在测试集上进行预测。如果Spearman相关性显著(p > 0.05;FDR < 0.05),则特征被认为是真正的阳性(TPs)。
对于预测指标“基因集”,编译了多个基因集,包括Hallmarks、Kegg、Reactome和PROGENy。为了识别原始单核多组学数据集中的相关基因集,使用decoupler-py的ULM方法在细胞水平推断基因集富集分数,并根据显著性进行筛选(最小细胞比例 = 20%;富集分数 > 0;BH FDR < 0.01)。对于GRN中的每个TF,执行单侧Fisher精确检验以确定其目标中哪些基因集过度表示(BH FDR < 0.01)。如果基因集在GRN中的至少一个TF调节子内过度表示,并且在原始数据集中也显著,则认为该基因集是一个TP。
对于文献衍生的“TF标记”指标,从人类蛋白质图谱(HPA)和TF-Marker数据库中检索数据。如果蛋白质在Lambert中被注释为TF,具有蛋白质水平的证据,其亚细胞位置包含“核”一词,并且不是在组织、细胞系或单细胞簇中非特异性表达,则保留HPA中的条目。两个数据库都根据与正在评估的数据集相关的术语进一步筛选。如果TF同时出现在GRN和数据库中,则认为该TF是一个真正的阳性(TP)。
对于文献衍生的“TF-TF对”指标,从IntAct中提取蛋白质-蛋白质相互作用。如果两个蛋白质都被注释为Lambert中的TFs,并且具有高置信度分数(MIscore > 0.75),则保留这些相互作用。对于给定的GRN,通过测试它们的靶基因是否相对于彼此过度表示来识别TF-TF对,使用单侧Fisher精确检验(BH FDR < 0.01)。如果TF-TF对同时出现在GRN和数据库中,则认为它是一个真正的阳性(TP)。
对于文献衍生的“TF结合”指标,我们处理了三个大型ChIP-seq测量数据库:ChIP-Atlas、Remap2022和UniBind。从ChIP-Atlas中,检索了在Lambert注释的TFs中显著的ChIP-seq TF结合峰(BH FDR < 1e-50;最大峰长 < 750)。从Remap2022中,检索了在Lambert注释的TFs中非冗余的ChIP-seq TF结合峰(最大峰长 < 750)。从UniBind中,检索了在Lambert注释的TFs中来自稳健集合的ChIP-seq TF结合峰(最大峰长 < 750)。对于每个数据库,使用bedtools合并了来自同一TF的重叠峰。对于给定的数据集和GRN,TF结合数据库被筛选为仅包括与原始染色质可及性数据中测量的CREs重叠的ChIP-seq峰,使用pyranges(版本0.1.2),原始基因表达数据中表达的TFs,以及与正在评估的数据集相关的细胞类型或组织术语。如果TF结合峰与GRN中该TF调控的CRE重叠,则视为TP。
对于文献衍生的“CREs”指标,处理了两个增强子注释(ENCODE和Zhang)、一个进化保守性(PhastCons)和一个SNP-trait(GWAS目录)数据库。此外,通过在Ensembl注释的TSS上打开基因组窗口(TSS起±1kb)生成了一个启动子资源。对于给定的数据集和GRN,CRE数据库被筛选为仅包括与原始染色质可及性数据中测量的CREs重叠的区域,使用pyranges(版本0.1.2)。如果CRE与GRN中的CRE重叠,则视为TP。
对于文献衍生的“基因”指标,处理了eQTL目录。如果基因在Ensembl中注释,并且链接显著(FDR < 1e-5),则保留CRE-基因相互作用。对于给定的数据集和GRN,数据库被筛选为仅包括在原始基因表达数据中表达的基因,以及与正在评估的数据集相关的细胞类型或组织术语。如果CRE-基因链接在GRN和数据集中都存在,则视为TP。
8.统计检验

相关文章:
GRN前沿:GRETA:从多模式单细胞数据推断基因调控网络方法的比较与评价
1.论文原名:Comparison and evaluation of methods to infer gene regulatory networks frommultimodal single-cell data 2.发表日期:20254.12.21 摘要: 细胞通过基因表达调节其功能,由转录因子和其他调节机制的复杂相互作用驱…...

python基础入门:4.4模块与包管理
Python模块与包管理完全指南:构建可维护的代码结构 # 示例项目结构 """ my_package/ ├── __init__.py ├── core/ │ ├── __init__.py │ ├── utils.py │ └── calculator.py ├── data/ │ └── config.json └── tes…...

《XSS跨站脚本攻击》
一、XSS简介 XSS全称(Cross Site Scripting)跨站脚本攻击,为了避免和CSS层叠样式表名称冲突,所以改为了XSS,是最常见的Web应用程序安全漏洞之一,位于OWASP top 10 2013/2017年度分别为第三名和第七名&…...
LC-两数之和、字母异位词分组、最长连续序列、移动零、盛最多水的容器
两数之和 class Solution {public int[] twoSum(int[] nums, int target) {int n nums.length; // 获取数组 nums 的长度// 外层循环:遍历数组中的每一个元素 nums[i]for(int i 0; i < n; i) {// 内层循环:从 nums[i] 的下一个元素 nums[j] 开始遍…...

Netty源码解析之线程池的实现(二):创建线程与执行任务
前言 先看下面的代码: public class MyTest {public static void main(String[] args) {//创建NioEventLoopGroupNioEventLoopGroup loopGroup new NioEventLoopGroup(3);System.out.println(Thread.currentThread()"准备执行任务");//执行任务for (in…...

IDEA - 一个启动类多次启动方法
More Run/Debug -> Modify Run Configuration -> modify options -> Allow mutiple instances...

U3D支持webgpu阅读
https://docs.unity3d.com/6000.1/Documentation/Manual/WebGPU-features.html 这里看到已经该有的差不多都有了 WOW VFX更是好东西 https://unity.com/cn/features/visual-effect-graph 这玩意儿化简了纯手搓一个特效的流程 如果按原理说就是compute shader刷position&#…...

C++广度优先搜索
简介 老规矩,先来介绍一下什么是广度优先搜索(至于这么长时间没更新是为什么,我放在文章结尾了,感兴趣可以看看,以后也是如此) 广度优先搜索,从名字就能听出来,他和深度优先搜索关…...

SVN 提交与原有文件类型不一样的文件时的操作
SVN 提交与原有文件类型不一样的文件时的操作 背景 SVN 服务器上原本的文件是软链接类型的,但是我将它改成普通文件再上传。出现了以下提示: 解决过程 本来想着通过 svn rm 和 svn add 来解决,但是行不通。 最终解决方案 svn rm --keep-…...
活动预告 | Power Hour: Copilot 引领商业应用的未来
课程介绍 智能化时代,商业应用如何实现突破?微软全球副总裁 Charles Lamanna 将为您深度解析,剖析其中关键因素。 在本次线上研讨会中,Charles Lamanna 将分享他在增强商业运营方面的独到见解与实战策略,深度解读商业…...
WPF 进度条(ProgressBar)示例一
本文讲述:WPF 进度条(ProgressBar)简单的样式修改和使用。 进度显示界面:使用UserControl把ProgressBar和进度值以及要显示的内容全部组装在UserControl界面中,方便其他界面直接进行使用。 <UserControl x:Class"DefProcessBarDemo…...

【C#】任务调度的实现原理与组件应用Quartz.Net
Quartz 是一个流行的开源作业调度库,最初由 Terracotta 开发,现在由 Terracotta 的一部分 Oracle 所有。它主要用于在 Java 应用程序中调度作业的执行。Quartz 使用了一种复杂的底层算法来管理任务调度,其中包括任务触发、执行、持久化以及集…...

UV - Python 包管理
文章目录 创建 uv 项目已有项目已有uv项目 创建 uv 项目 # 创建项目 uv init m3 # 创建环境 cd m3 uv venv --python 3.11 # 激活环境 source .venv/bin/activate # 添加库 uv add flask 如果创建项目后,给库取别的名字,add 的时候,会…...

pytorch torch.linalg模块介绍
torch.linalg 是 PyTorch 的 线性代数 (Linear Algebra) 子模块,它提供了许多 高效的矩阵操作和分解方法,类似于 NumPy 的 numpy.linalg 或 SciPy 的 scipy.linalg,但针对 GPU 加速和自动微分 进行了优化。 1. 矩阵基本运算 矩阵乘法 torc…...

光伏-报告显示,假期内,硅料端签单顺序发货相对稳定。若3月份下游存提产,则不排除硅料价格有上调预期。
据TrendForce集邦咨询报告显示,假期内,硅料端按照前期签单顺序发货,相对稳定。若3月份下游存提产,则不排除硅料价格有上调预期。 002306中科云网 旅游 | 公司为提供复合菜系特色餐饮的连锁企业,形成了以粤菜ÿ…...

【web自动化】指定chromedriver以及chrome路径
selenium自动化,指定chromedriver,以及chrome路径 对应这篇文章,可以点击查看,详情 from selenium import webdriverdef get_driver():# 获取配置对象option webdriver.ChromeOptions()option.add_experimental_option("de…...
面试题及参考答案)
顺丰数据分析(数据挖掘)面试题及参考答案
你觉得数据分析人员必备的技能有哪些? 数据分析人员需具备多方面技能,以应对复杂的数据处理与解读工作。 数据处理能力:这是基础且关键的技能。数据常以杂乱、不完整的形式存在,需通过清洗,去除重复、错误及缺失值数据,确保数据质量。例如,在电商销售数据中,可能存在价…...

Android studio:顶部导航栏Toolbar
主流APP在顶部都配有导航栏,在 Android 中,ActionBar 是默认启用的,它是位于屏幕顶部的一个工具栏,用来放置应用的标题、导航和操作菜单。 如果你想使用自定义的 Toolbar 来替代 ActionBar,应该先关闭它。可以通过设置…...

mmap 文件映射
🌈 个人主页:Zfox_ 🔥 系列专栏:Linux 目录 一:🔥 mmap介绍🦋 基本说明🦋 参数介绍🦋 返回值 二:🔥 demo代码🦋 写入映射🦋…...

基于微信小程序的医院预约挂号系统的设计与实现
hello hello~ ,这里是 code袁~💖💖 ,欢迎大家点赞🥳🥳关注💥💥收藏🌹🌹🌹 🦁作者简介:一名喜欢分享和记录学习的在校大学生…...

3个关键功能解析:USBToolBox如何简化macOS与Windows的USB端口映射难题
3个关键功能解析:USBToolBox如何简化macOS与Windows的USB端口映射难题 【免费下载链接】tool the USBToolBox tool 项目地址: https://gitcode.com/gh_mirrors/too/tool 在Hackintosh和跨平台开发领域,USB端口映射一直是个令人头疼的技术难题。US…...

本地柴油发电机组排行2023年最新榜单
柴油发电机是通过燃烧柴油驱动发动机,进而发电的设备,广泛应用于电力中断或无电网地区。1. 柴油发电机的核心工作原理是什么?柴油发电机是一种将化学能转化为电能的设备,其核心是柴油发动机与交流发电机的组合。当柴油在发动机内燃…...

番茄小说下载器终极指南:三步构建你的离线阅读自由王国
番茄小说下载器终极指南:三步构建你的离线阅读自由王国 【免费下载链接】Tomato-Novel-Downloader 番茄小说下载器不精简版 项目地址: https://gitcode.com/gh_mirrors/to/Tomato-Novel-Downloader 你是否曾在地铁里读到精彩章节时突然断网?是否在…...

服务器日志分析实战:用Python追踪HTTP 404错误并可视化异常频率
作为一名爬虫开发者或网站运维人员,服务器日志就像飞机的“黑匣子”——它记录了每个请求的来龙去脉。而404错误(页面未找到)尤其值得关注:它可能是用户输错了网址,可能是你爬虫的URL构造逻辑有漏洞,也可能是网站改版后旧的链接失效了。更严重的是,大量突然涌出的404请求…...

基于双T振荡器的正弦波LED调光电路设计与实践
1. 项目概述:用双T振荡器实现正弦波LED调光最近在捣鼓一些氛围灯项目,总感觉用单片机PWM做的呼吸灯效果有点“硬”,那种线性的明暗变化看久了难免审美疲劳。于是翻出以前模拟电路的老本行,琢磨着能不能用纯硬件的方式,…...

MongoDB Limit 与 Skip 方法详解
MongoDB Limit 与 Skip 方法详解 引言 MongoDB 是一个高性能、可伸缩的文档存储系统,它提供了强大的数据存储和查询功能。在处理大量数据时,Limit 与 Skip 方法是 MongoDB 中常用的查询优化工具。本文将详细介绍 MongoDB 中的 Limit 与 Skip 方法,包括其基本用法、性能影响…...

告别CAJ格式困扰:3分钟学会用开源工具将知网文献转为PDF
告别CAJ格式困扰:3分钟学会用开源工具将知网文献转为PDF 【免费下载链接】caj2pdf Convert CAJ (China Academic Journals) files to PDF. 转换中国知网 CAJ 格式文献为 PDF。佛系转换,成功与否,皆是玄学。 项目地址: https://gitcode.com/…...

关联规则挖掘在Calabi-Yau流形Hodge数分析中的应用与复现
1. 项目概述:当数据挖掘遇见高维几何在理论物理和代数几何的交叉领域,Calabi-Yau流形一直扮演着核心角色。这些具有特殊拓扑结构的空间,不仅是弦理论中额外维度紧化的关键候选者,其本身丰富的数学性质也吸引着无数研究者。然而&am…...

终极歌词同步神器LRCGET:5分钟为你的音乐库添加完美歌词
终极歌词同步神器LRCGET:5分钟为你的音乐库添加完美歌词 【免费下载链接】lrcget Utility for mass-downloading LRC synced lyrics for your offline music library. 项目地址: https://gitcode.com/gh_mirrors/lr/lrcget 你是否厌倦了在听歌时手动搜索歌词…...

3分钟掌握JetBrains IDE试用期重置:终极完整指南
3分钟掌握JetBrains IDE试用期重置:终极完整指南 【免费下载链接】ide-eval-resetter 项目地址: https://gitcode.com/gh_mirrors/id/ide-eval-resetter JetBrains IDE试用期重置工具(ide-eval-resetter)是一个开源项目,专…...